
Polyamine Homeostasis in Development and Disease

Abstract
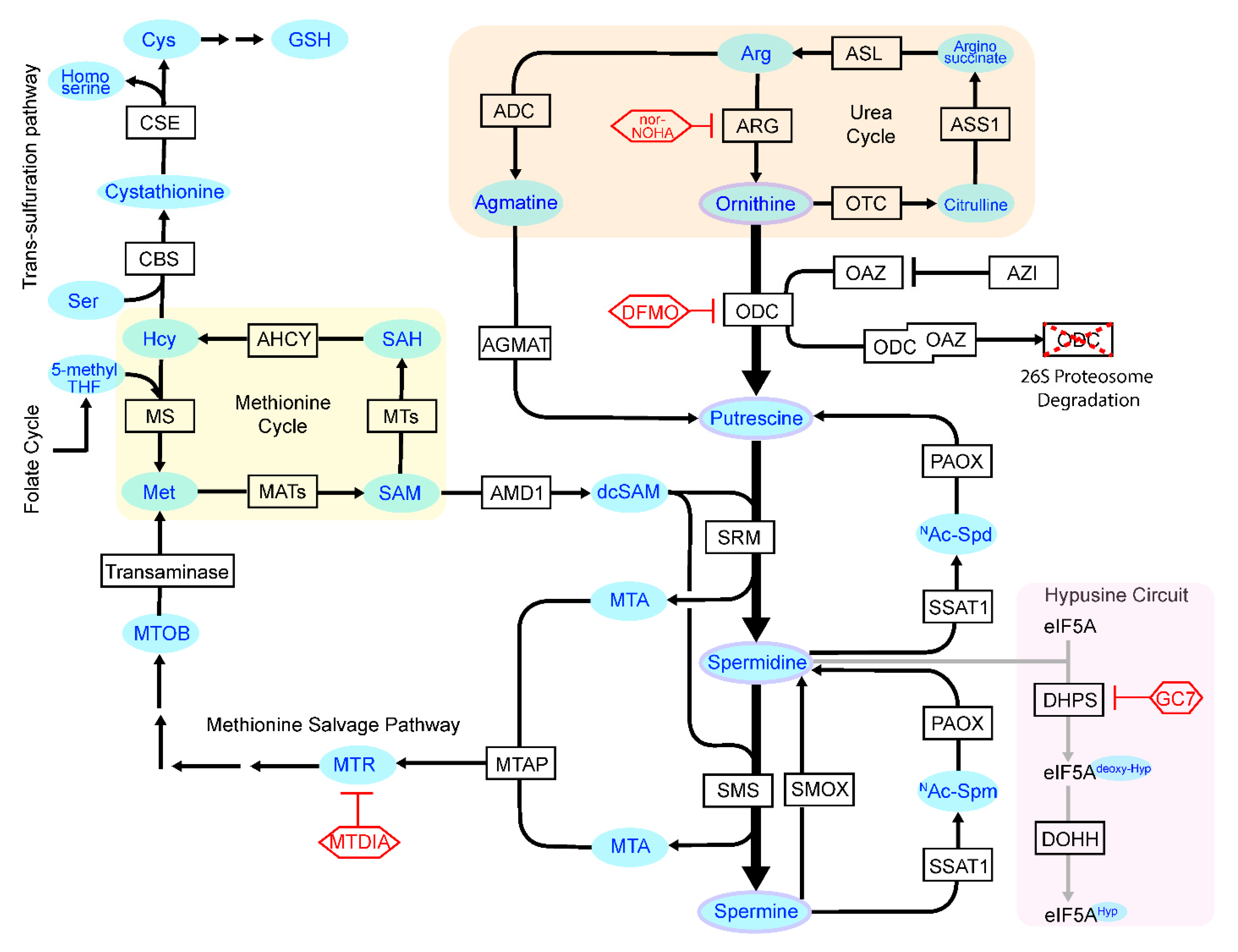
:1. Introduction

2. Beneficial Aspects of Polyamines and Effects of Polyamine Loss on Health
2.1. Spermidine and Spermine Can Extend Lifespan
2.2. Cardioprotective Effects of Spermidine
2.3. Neuroprotective Effects of Polyamines
3. Detrimental Aspects of Polyamines: Effects of High Polyamine Levels
3.1. Control of ODC
3.2. Regulation of AMD1
3.3. Control of Polyamine Catabolism and Transport
3.4. Control of Polyamine Homeostasis and Interacting Metabolic Pathways in Cancer

{kind=link}
| Interacting Metabolic Pathway | Deregulated Enzymes/Genes | Target (Inhibitor) | Target Polyamine (Inhibitor/Compound) | Description | Disease | Ref. |
|---|---|---|---|---|---|---|
| Arginine pathway | ASS1 | Arg depletion (ADI-PEG20) | ODC inhibition (DFMO) | ASS1-deficient cells have decreased levels of acetylated polyamines along with compensatory increases in polyamine biosynthetic enzymes. | Malignant pleural mesothelioma (MPM) | [130] |
| Arginine pathway/Urea cycle | ARG2 | N/A | Polyamine toxicity | ARG2 suppresses tumor growth via depletion of biosynthetic cofactor PLP and toxic polyamine accumulation. | Clear cell renal cell carcinoma (ccRCC) | [131] |
| Urea cycle | p53 repressive target genes; CPS1, OTC and ARG1 | N/A | ODC translation | p53-induced ammonia accumulation represses ODC translation. | Colon cancer | [63] |
| Arginine pathway/Urea cycle | ARG1 | Inhibition of arginase (nor-NOHA) | ODC (DFMO) | Increased polyamine production in PP6-deficient keratinocytes facilitates self-RNA sensing by dendritic cells in psoriasis. | Psoriasis | [132] |
| Cysteine metabolism | Cysteine starvation | MTAP deletion | MTAP deletion upregulates polyamine pathway, which promotes ferroptosis under cysteine starvation. | Colorectal, breast and pancreatic cancers, and glioblastoma | [119] | |
| Methionine salvage pathway | MTAP inhibition (MTDIA) | SSAT activation (BENSpm) | While keeping the high polyamines flux, SAM pools are depleted by inhibition of methionine salvage pathway. | Prostate cancers | [115] |
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines. Annu. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hall, J. Endogenous polyamine function—The RNA perspective. Nucleic Acids Res. 2014, 42, 11275–11290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, A.; Caldarera, C.M.; Giordano, E. Chromatin remodeling by polyamines and polyamine analogs. Amino Acids 2014, 46, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U. Naturally occurring polyamines: Interaction with macromolecules. Curr. Protein Pept. Sci. 2005, 6, 559–566. [Google Scholar] [CrossRef]
- Çelik, V.K.; Kapancık, S.; Kaçan, T.; Kaçan, S.B.; Kapancık, S.; Kılıçgün, H. Serum levels of polyamine synthesis enzymes increase in diabetic patients with breast cancer. Endocr. Connect. 2017, 6, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Dallmann, K.; Junker, H.; Balabanov, S.; Zimmermann, U.; Giebel, J.; Walther, R. Human agmatinase is diminished in the clear cell type of renal cell carcinoma. Int. J. Cancer. 2004, 108, 342–347. [Google Scholar] [CrossRef]
- Li, G.; Regunathan, S.; Barrow, C.J.; Eshraghi, J.; Cooper, R.; Reis, D.J. Agmatine: An endogenous clonidine-displacing substance in the brain. Science 1994, 263, 966–969. [Google Scholar] [CrossRef]
- Mistry, S.K.; Burwell, T.J.; Chambers, R.M.; Rudolph-Owen, L.; Spaltmann, F.; Cook, W.J.; Morris, S.M., Jr. Cloning of human agmatinase. An alternate path for polyamine synthesis induced in liver by hepatitis B virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G375–G381. [Google Scholar] [CrossRef]
- Zhu, H.E.; Yin, J.Y.; Chen, D.X.; He, S.; Chen, H. Agmatinase promotes the lung adenocarcinoma tumorigenesis by activating the NO-MAPKs-PI3K/Akt pathway. Cell Death Dis. 2019, 10, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, A.E. Mammalian polyamine metabolism and function. IUBMB Life 2009, 61, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Matsufuji, S.; Matsufuji, T.; Miyazaki, Y.; Murakami, Y.; Atkins, J.F.; Gesteland, R.F.; Hayashi, S. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 1995, 80, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Kitani, T.; Fujisawa, H. Purification and characterization of antizyme inhibitor of ornithine decarboxylase from rat liver. Biochim. Biophys. Acta 1989, 991, 44–49. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsufuji, S.; Nishiyama, M.; Hayashi, S. Properties and fluctuations in vivo of rat liver antizyme inhibitor. Biochem. J. 1989, 259, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shantz, L.M.; Holm, I.; Jänne, O.A.; Pegg, A.E. Regulation of S-adenosylmethionine decarboxylase activity by alterations in the intracellular polyamine content. Biochem. J. 1992, 288, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Bercovich, Z.; Tsvetkov, P.; Shaul, Y.; Kahana, C. 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 2005, 17, 645–655. [Google Scholar] [CrossRef]
- Iwami, K.; Wang, J.Y.; Jain, R.; McCormack, S.; Johnson, L.R. Intestinal ornithine decarboxylase: Half-life and regulation by putrescine. Am. J. Physiol. 1990, 258, G308–G315. [Google Scholar] [CrossRef]
- Zwighaft, Z.; Aviram, R.; Shalev, M.; Rousso-Noori, L.; Kraut-Cohen, J.; Golik, M.; Brandis, A.; Reinke, H.; Aharoni, A.; Kahana, C.; et al. Circadian Clock Control by Polyamine Levels through a Mechanism that Declines with Age. Cell Metab. 2015, 22, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Igarashi, K. Polyamines and their metabolites as diagnostic markers of human diseases. Biomol. Ther. 2013, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Auvinen, M.; Laine, A.; Paasinen-Sohns, A.; Kangas, A.; Kangas, L.; Saksela, O.; Andersson, L.C.; Hölttä, E. Human ornithine decarboxylase-overproducing NIH3T3 cells induce rapidly growing, highly vascularized tumors in nude mice. Cancer Res. 1997, 57, 3016–3025. [Google Scholar]
- Clifford, A.; Morgan, D.; Yuspa, S.H.; Soler, A.P.; Gilmour, S. Role of ornithine decarboxylase in epidermal tumorigenesis. Cancer Res. 1995, 55, 1680–1686. [Google Scholar]
- Mamont, P.S.; Duchesne, M.C.; Grove, J.; Bey, P. Anti-proliferative properties of DL-α-difluoromethyl ornithine in cultured cells. A consequence of the irreversible inhibition of ornithine decarboxylase. Biochem. Biophys. Res. Commun. 1978, 81, 58–66. [Google Scholar] [CrossRef]
- Nilsson, J.A.; Keller, U.B.; Baudino, T.A.; Yang, C.; Norton, S.; Old, J.A.; Nilsson, L.M.; Neale, G.; Kramer, D.L.; Porter, C.W.; et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell 2005, 7, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rounbehler, R.J.; Li, W.; Hall, M.A.; Yang, C.; Fallahi, M.; Cleveland, J.L. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 2009, 69, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Sholler, G.L.S.; Ferguson, W.; Bergendahl, G.; Bond, J.P.; Neville, K.; Eslin, D.; Brown, V.; Roberts, W.; Wada, R.K.; Oesterheld, J.; et al. Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci. Rep. 2018, 8, 14445. [Google Scholar] [CrossRef] [Green Version]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrada-Gimenez, M.; Pietilä, M.; Loimas, S.; Pirinen, E.; Hyvönen, M.T.; Keinänen, T.A.; Jänne, J.; Alhonen, L. Continuous oxidative stress due to activation of polyamine catabolism accelerates aging and protects against hepatotoxic insults. Transgenic Res. 2011, 20, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Shiina, R.; Kashiwagi, K.; Igarashi, K. Decrease in polyamines with aging and their ingestion from food and drink. J. Biochem. 2006, 139, 81–90. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Gerner, E.W.; Bruckheimer, E.; Cohen, A. Cancer pharmacoprevention: Targeting polyamine metabolism to manage risk factors for colon cancer. J. Biol. Chem. 2018, 293, 18770–18778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [Green Version]
- Wallace, H.M.; Fraser, A.V. Inhibitors of polyamine metabolism: Review article. Amino Acids 2004, 26, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Akasaka, Y.; Ikegaya, H. Correlation of polyamines, acrolein-conjugated lysine and polyamine metabolic enzyme levels with age in human liver. Heliyon 2020, 6, e05031. [Google Scholar] [CrossRef]
- Holbert, C.E.; Dunworth, M.; Foley, J.R.; Dunston, T.T.; Stewart, T.M.; Casero, R.A., Jr. Autophagy induction by exogenous polyamines is an artifact of bovine serum amine oxidase activity in culture serum. J. Biol. Chem. 2020, 295, 9061–9068. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Becker, L.; Ang, L.C.; Kish, S.J. Polyamines in human brain: Regional distribution and influence of aging. J. Neurochem. 1995, 65, 636–642. [Google Scholar] [CrossRef]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 2013, 16, 1453–1460. [Google Scholar] [CrossRef]
- Gupta, V.K.; Pech, U.; Bhukel, A.; Fulterer, A.; Ender, A.; Mauermann, S.F.; Andlauer, T.F.; Antwi-Adjei, E.; Beuschel, C.; Thriene, K.; et al. Spermidine Suppresses Age-Associated Memory Impairment by Preventing Adverse Increase of Presynaptic Active Zone Size and Release. PLoS Biol. 2016, 14, e1002563. [Google Scholar] [CrossRef] [PubMed]
- Maglione, M.; Kochlamazashvili, G.; Eisenberg, T.; Rácz, B.; Michael, E.; Toppe, D.; Stumpf, A.; Wirth, A.; Zeug, A.; Müller, F.E.; et al. Spermidine protects from age-related synaptic alterations at hippocampal mossy fiber-CA3 synapses. Sci. Rep. 2019, 9, 19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Muller, M.; et al. Dietary spermidine improves cognitive function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef]
- Snyder, R.D.; Robinson, A. Recessive sex-linked mental retardation in the absence of other recognizable abnormalities. Report of a family. Clin. Pediatr. 1969, 8, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.E.; Wang, X.; Stevenson, R.E.; Pegg, A.E. Spermine synthase deficiency resulting in X-linked intellectual disability (Snyder-Robinson syndrome). Methods Mol. Biol. 2011, 720, 437–445. [Google Scholar] [CrossRef]
- Murray-Stewart, T.; Dunworth, M.; Foley, J.R.; Schwartz, C.E.; Casero, R.A., Jr. Polyamine Homeostasis in Snyder-Robinson Syndrome. Med. Sci. 2018, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Albert, T.; Urlbauer, B.; Kohlhuber, F.; Hammersen, B.; Eick, D. Ongoing mutations in the N-terminal domain of c-Myc affect transactivation in Burkitt’s lymphoma cell lines. Oncogene 1994, 9, 759–763. [Google Scholar]
- Li, C.; Brazill, J.M.; Liu, S.; Bello, C.; Zhu, Y.; Morimoto, M.; Cascio, L.; Pauly, R.; Diaz-Perez, Z.; Malicdan, M.C.V.; et al. Spermine synthase deficiency causes lysosomal dysfunction and oxidative stress in models of Snyder-Robinson syndrome. Nat. Commun. 2017, 8, 1257. [Google Scholar] [CrossRef] [PubMed]
- Matheis, F.; Muller, P.A.; Mucida, D. Gut macrophages: Key players in intestinal immunity and tissue physiology. Curr. Opin. Immunol. 2020, 62, 54–61. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [Green Version]
- Shantz, L.M.; Levin, V.A. Regulation of ornithine decarboxylase during oncogenic transformation: Mechanisms and therapeutic potential. Amino Acids 2007, 33, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, A.S.; Geerts, D. Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem. 2018, 293, 18757–18769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, A.T.; Hogarty, M.D. Myc, Oncogenic Protein Translation, and the Role of Polyamines. Med. Sci. 2018, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Origanti, S.; Nowotarski, S.L.; Carr, T.D.; Sass-Kuhn, S.; Xiao, L.; Wang, J.Y.; Shantz, L.M. Ornithine decarboxylase mRNA is stabilized in an mTORC1-dependent manner in Ras-transformed cells. Biochem. J. 2012, 442, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.P.; Loughran, G.; Atkins, J.F. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc. Natl. Acad. Sci. USA 2008, 105, 10079–10084. [Google Scholar] [CrossRef] [Green Version]
- Pyronnet, S.; Pradayrol, L.; Sonenberg, N. A cell cycle-dependent internal ribosome entry site. Mol. Cell 2000, 5, 607–616. [Google Scholar] [CrossRef]
- Sammons, M.A.; Antons, A.K.; Bendjennat, M.; Udd, B.; Krahe, R.; Link, A.J. ZNF9 activation of IRES-mediated translation of the human ODC mRNA is decreased in myotonic dystrophy type 2. PLoS ONE 2010, 5, e9301. [Google Scholar] [CrossRef]
- D’Amico, D.; Antonucci, L.; Di Magno, L.; Coni, S.; Sdruscia, G.; Macone, A.; Miele, E.; Infante, P.; Di Marcotullio, L.; De Smaele, E.; et al. Non-canonical Hedgehog/AMPK-Mediated Control of Polyamine Metabolism Supports Neuronal and Medulloblastoma Cell Growth. Dev. Cell 2015, 35, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Coffino, P. Regulation of cellular polyamines by antizyme. Nat. Rev. Mol. Cell Biol. 2001, 2, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Chen, S.F.; Hsieh, J.Y.; Chou, F.; Wang, Y.H.; Lin, W.T.; Lee, P.Y.; Yu, Y.J.; Lin, L.Y.; Lin, T.S.; et al. Structural basis of antizyme-mediated regulation of polyamine homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Kahana, C. Antizyme and antizyme inhibitor, a regulatory tango. Cell Mol. Life Sci. 2009, 66, 2479–2488. [Google Scholar] [CrossRef]
- Ivanov, I.P.; Atkins, J.F. Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: Close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. 2007, 35, 1842–1858. [Google Scholar] [CrossRef]
- Kahana, C. The antizyme family for regulating polyamines. J. Biol. Chem. 2018, 293, 18730–18735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.P.; Shin, B.S.; Loughran, G.; Tzani, I.; Young-Baird, S.K.; Cao, C.; Atkins, J.F.; Dever, T.E. Polyamine Control of Translation Elongation Regulates Start Site Selection on Antizyme Inhibitor mRNA via Ribosome Queuing. Mol. Cell 2018, 70, 254–264.e256. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Coffino, P. Degradation of ornithine decarboxylase: Exposure of the C-terminal target by a polyamine-inducible inhibitory protein. Mol. Cell. Biol. 1993, 13, 2377–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bupp, C.P.; Schultz, C.R.; Uhl, K.L.; Rajasekaran, S.; Bachmann, A.S. Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features. Am. J. Med. Genet. A 2018, 176, 2548–2553. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.R.; Bupp, C.P.; Rajasekaran, S.; Bachmann, A.S. Biochemical features of primary cells from a pediatric patient with a gain-of-function ODC1 genetic mutation. Biochem. J. 2019, 476, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Geerts, D.; Feith, D.J.; Mocz, G.; Koster, J.; Bachmann, A.S. Novel interaction of ornithine decarboxylase with sepiapterin reductase regulates neuroblastoma cell proliferation. J. Mol. Biol. 2014, 426, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Yco, L.P.; Geerts, D.; Mocz, G.; Koster, J.; Bachmann, A.S. Effect of sulfasalazine on human neuroblastoma: Analysis of sepiapterin reductase (SPR) as a new therapeutic target. BMC Cancer 2015, 15, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, A.E. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009, 46, 25–45. [Google Scholar] [CrossRef] [Green Version]
- Law, G.L.; Raney, A.; Heusner, C.; Morris, D.R. Polyamine regulation of ribosome pausing at the upstream open reading frame of S-adenosylmethionine decarboxylase. J. Biol. Chem. 2001, 276, 38036–38043. [Google Scholar] [CrossRef]
- Yordanova, M.M.; Loughran, G.; Zhdanov, A.V.; Mariotti, M.; Kiniry, S.J.; O’Connor, P.B.F.; Andreev, D.E.; Tzani, I.; Saffert, P.; Michel, A.M.; et al. AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nature 2018, 553, 356–360. [Google Scholar] [CrossRef]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martin-Martin, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef]
- Su, C.Y.; Chang, Y.C.; Chan, Y.C.; Lin, T.C.; Huang, M.S.; Yang, C.J.; Hsiao, M. MTAP is an independent prognosis marker and the concordant loss of MTAP and p16 expression predicts short survival in non-small cell lung cancer patients. Eur. J. Surg. Oncol. 2014, 40, 1143–1150. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, L.; Thiagalingam, A.; Nelkin, B.D.; Casero, R.A., Jr. The identification of a cis-element and a trans-acting factor involved in the response to polyamines and polyamine analogues in the regulation of the human spermidine/spermine N1-acetyltransferase gene transcription. J. Biol. Chem. 1998, 273, 34623–34630. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Devereux, W.; Stewart, T.M.; Casero, R.A., Jr. Cloning and characterization of human polyamine-modulated factor-1, a transcriptional cofactor that regulates the transcription of the spermidine/spermine N(1)-acetyltransferase gene. J. Biol. Chem. 1999, 274, 22095–22101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Devereux, W.; Stewart, T.M.; Casero, R.A., Jr. Characterization of the interaction between the transcription factors human polyamine modulated factor (PMF-1) and NF-E2-related factor 2 (Nrf-2) in the transcriptional regulation of the spermidine/spermine N1-acetyltransferase (SSAT) gene. Biochem. J. 2001, 355, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Butcher, N.J.; Broadhurst, G.M.; Minchin, R.F. Polyamine-dependent regulation of spermidine-spermine N1-acetyltransferase mRNA translation. J. Biol. Chem. 2007, 282, 28530–28539. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.P.; Atkins, J.F.; Michael, A.J. A profusion of upstream open reading frame mechanisms in polyamine-responsive translational regulation. Nucleic Acids Res. 2010, 38, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Perez-Leal, O.; Barrero, C.A.; Clarkson, A.B.; Casero, R.A., Jr.; Merali, S. Polyamine-regulated translation of spermidine/spermine-N1-acetyltransferase. Mol. Cell. Biol. 2012, 32, 1453–1467. [Google Scholar] [CrossRef] [Green Version]
- Casero, R.A., Jr.; Celano, P.; Ervin, S.J.; Wiest, L.; Pegg, A.E. High specific induction of spermidine/spermine N1-acetyltransferase in a human large cell lung carcinoma. Biochem. J. 1990, 270, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Murray-Stewart, T.R.; Woster, P.M.; Casero, R.A., Jr. Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 2016, 473, 2937–2953. [Google Scholar] [CrossRef] [Green Version]
- Pledgie-Tracy, A.; Billam, M.; Hacker, A.; Sobolewski, M.D.; Woster, P.M.; Zhang, Z.; Casero, R.A.; Davidson, N.E. The role of the polyamine catabolic enzymes SSAT and SMO in the synergistic effects of standard chemotherapeutic agents with a polyamine analogue in human breast cancer cell lines. Cancer Chemother. Pharm. 2010, 65, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Creaven, P.J.; Perez, R.; Pendyala, L.; Meropol, N.J.; Loewen, G.; Levine, E.; Berghorn, E.; Raghavan, D. Unusual central nervous system toxicity in a phase I study of N1N11 diethylnorspermine in patients with advanced malignancy. Investig. New Drugs 1997, 15, 227–234. [Google Scholar] [CrossRef]
- Hahm, H.A.; Ettinger, D.S.; Bowling, K.; Hoker, B.; Chen, T.L.; Zabelina, Y.; Casero, R.A., Jr. Phase I study of N(1),N(11)-diethylnorspermine in patients with non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 684–690. [Google Scholar]
- Streiff, R.R.; Bender, J.F. Phase 1 study of N1-N11-diethylnorspermine (DENSPM) administered TID for 6 days in patients with advanced malignancies. Investig. New Drugs 2001, 19, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Armstrong, D.K.; Fetting, J.H.; Carducci, M.K.; Riley, C.D.; Bender, J.F.; Casero, R.A., Jr.; Davidson, N.E. A Phase II study of the polyamine analog N1,N11-diethylnorspermine (DENSpm) daily for five days every 21 days in patients with previously treated metastatic breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 5922–5928. [Google Scholar]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, A.C.; Shields, C.E.D.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, R.; Asim, M.; Piazuelo, M.B.; Yan, F.; Barry, D.P.; Sierra, J.C.; Delgado, A.G.; Hill, S.; Casero, R.A., Jr.; Bravo, L.E.; et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology 2014, 146, 1739–1751 e1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, A.C.; Jadallah, S.; Toubaji, A.; Lecksell, K.; Hicks, J.L.; Kowalski, J.; Bova, G.S.; De Marzo, A.M.; Netto, G.J.; Casero, R.A., Jr. Increased spermine oxidase expression in human prostate cancer and prostatic intraepithelial neoplasia tissues. Prostate 2008, 68, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Sun, D.; Zhang, J.; Xue, R.; Janssen, H.L.A.; Tang, W.; Dong, L. Spermine oxidase is upregulated and promotes tumor growth in hepatocellular carcinoma. Hepatol. Res. 2018, 48, 967–977. [Google Scholar] [CrossRef]
- Chaturvedi, R.; de Sablet, T.; Asim, M.; Piazuelo, M.B.; Barry, D.P.; Verriere, T.G.; Sierra, J.C.; Hardbower, D.M.; Delgado, A.G.; Schneider, B.G.; et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 2015, 34, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray-Stewart, T.; Sierra, J.C.; Piazuelo, M.B.; Mera, R.M.; Chaturvedi, R.; Bravo, L.E.; Correa, P.; Schneider, B.G.; Wilson, K.T.; Casero, R.A. Epigenetic silencing of miR-124 prevents spermine oxidase regulation: Implications for Helicobacter pylori-induced gastric cancer. Oncogene 2016, 35, 5480–5488. [Google Scholar] [CrossRef] [Green Version]
- Sierra, J.C.; Piazuelo, M.B.; Luis, P.B.; Barry, D.P.; Allaman, M.M.; Asim, M.; Sebrell, T.A.; Finley, J.L.; Rose, K.L.; Hill, S.; et al. Spermine oxidase mediates Helicobacter pylori-induced gastric inflammation, DNA damage, and carcinogenic signaling. Oncogene 2020, 39, 4465–4474. [Google Scholar] [CrossRef]
- Igarashi, K.; Uemura, T.; Kashiwagi, K. Acrolein: An Effective Biomarker for Tissue Damage Produced from Polyamines. Methods Mol. Biol 2018, 1694, 459–468. [Google Scholar] [CrossRef]
- Tomitori, H.; Usui, T.; Saeki, N.; Ueda, S.; Kase, H.; Nishimura, K.; Kashiwagi, K.; Igarashi, K. Polyamine oxidase and acrolein as novel biochemical markers for diagnosis of cerebral stroke. Stroke 2005, 36, 2609–2613. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki, T.; Ko, K.; Watanabe, K.; Dohmae, N.; Sakamoto, A.; Terui, Y.; Toida, T.; Kashiwagi, K.; Igarashi, K. Inhibition of dendritic spine extension through acrolein conjugation with α-, β-tubulin proteins. Int. J. Biochem. Cell Biol. 2019, 113, 58–66. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki, T.; Ko, K.; Nakamura, M.; Dohmae, N.; Sakamoto, A.; Terui, Y.; Toida, T.; Kashiwagi, K.; Igarashi, K. Structural change and degradation of cytoskeleton due to the acrolein conjugation with vimentin and actin during brain infarction. Cytoskeleton 2020, 77, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Uemura, T.; Kashiwagi, K. Assessing acrolein for determination of the severity of brain stroke, dementia, renal failure, and Sjögren’s syndrome. Amino Acids 2020, 52, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Takasaka, T.; Igarashi, K.; Ikegaya, H. Spermine oxidase promotes bile canalicular lumen formation through acrolein production. Sci. Rep. 2017, 7, 14841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, N.M.; Ju, S.; Verbitsky, M.; Ross, B.; Geddie, M.L.; Rockenstein, E.; Adame, A.; Muhammad, A.; Vonsattel, J.P.; Ringe, D.; et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16970–16975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; D’Amelio, M.; Cavallucci, V.; Moreno, S.; Berger, J.; Nardacci, R.; Marcoli, M.; Maura, G.; Piacentini, M.; et al. A New Transgenic Mouse Model for Studying the Neurotoxicity of Spermine Oxidase Dosage in the Response to Excitotoxic Injury. PLoS ONE 2013, 8, e64810. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Pietropaoli, S.; Leonetti, A.; Cervetto, C.; Venturini, A.; Mastrantonio, R.; Baroli, G.; Persichini, T.; Colasanti, M.; Maura, G.; Marcoli, M.; et al. Glutamate Excitotoxicity Linked to Spermine Oxidase Overexpression. Mol. Neurobiol. 2018, 55, 7259–7270. [Google Scholar] [CrossRef]
- Cervetto, C.; Vergani, L.; Passalacqua, M.; Ragazzoni, M.; Venturini, A.; Cecconi, F.; Berretta, N.; Mercuri, N.; D’Amelio, M.; Maura, G.; et al. Astrocyte-Dependent Vulnerability to Excitotoxicity in Spermine Oxidase-Overexpressing Mouse. Neuromol. Med. 2016, 18, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Baroli, G.; Fratini, E.; Pietropaoli, S.; Marcoli, M.; Mariottini, P.; Cervelli, M. Epileptic seizures and oxidative stress in a mouse model over-expressing spermine oxidase. Amino Acids 2020, 52, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Affronti, H.C.; Foster, B.A.; Karasik, E.; Gillard, B.; Morrison, C.; Mohler, J.; Phillips, J.G.; Smiraglia, D.J. The essential role of methylthioadenosine phosphorylase in prostate cancer. Oncotarget 2016, 7, 14380–14393. [Google Scholar] [CrossRef] [Green Version]
- Affronti, H.C.; Rowsam, A.M.; Pellerite, A.J.; Rosario, S.R.; Long, M.D.; Jacobi, J.J.; Bianchi-Smiraglia, A.; Boerlin, C.S.; Gillard, B.M.; Karasik, E.; et al. Pharmacological polyamine catabolism upregulation with methionine salvage pathway inhibition as an effective prostate cancer therapy. Nat. Commun. 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, R.J.; Neims, A.H.; McManis, J.S.; Hawthorne, T.R.; Vinson, J.R.; Bortell, R.; Ingeno, M.J. Synthetic polyamine analogues as antineoplastics. J. Med. Chem. 1988, 31, 1183–1190. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Celano, P.; Ervin, S.J.; Porter, C.W.; Bergeron, R.J.; Libby, P.R. Differential induction of spermidine/spermine N1-acetyltransferase in human lung cancer cells by the bis(ethyl)polyamine analogues. Cancer Res. 1989, 49, 3829–3833. [Google Scholar]
- Porter, C.W.; Bernacki, R.J.; Miller, J.; Bergeron, R.J. Antitumor activity of N1,N11-bis(ethyl)norspermine against human melanoma xenografts and possible biochemical correlates of drug action. Cancer Res. 1993, 53, 581–586. [Google Scholar]
- Zhang, T.; Bauer, C.; Newman, A.C.; Uribe, A.H.; Athineos, D.; Blyth, K.; Maddocks, O.D.K. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat. Metab. 2020, 2, 1062–1076. [Google Scholar] [CrossRef]
- Subhi, A.L.; Diegelman, P.; Porter, C.W.; Tang, B.; Lu, Z.J.; Markham, G.D.; Kruger, W.D. Methylthioadenosine phosphorylase regulates ornithine decarboxylase by production of downstream metabolites. J. Biol. Chem. 2003, 278, 49868–49873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subhi, A.L.; Tang, B.; Balsara, B.R.; Altomare, D.A.; Testa, J.R.; Cooper, H.S.; Hoffman, J.P.; Meropol, N.J.; Kruger, W.D. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7290–7296. [Google Scholar] [CrossRef] [Green Version]
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef]
- Huang, H.Y.; Wu, W.R.; Wang, Y.H.; Wang, J.W.; Fang, F.M.; Tsai, J.W.; Li, S.H.; Hung, H.C.; Yu, S.C.; Lan, J.; et al. ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: Aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2861–2872. [Google Scholar] [CrossRef] [Green Version]
- Bowles, T.L.; Kim, R.; Galante, J.; Parsons, C.M.; Virudachalam, S.; Kung, H.J.; Bold, R.J. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int. J. Cancer J. 2008, 123, 1950–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szlosarek, P.W.; Luong, P.; Phillips, M.M.; Baccarini, M.; Stephen, E.; Szyszko, T.; Sheaff, M.T.; Avril, N. Metabolic response to pegylated arginine deiminase in mesothelioma with promoter methylation of argininosuccinate synthetase. J. Clin. Oncol. 2013, 31, e111–e113. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Tsai, W.B.; Wangpaichitr, M.; Tsukamoto, T.; Savaraj, N.; Feun, L.G.; Kuo, M.T. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol. Cancer Ther. 2013, 12, 2581–2590. [Google Scholar] [CrossRef] [Green Version]
- Locke, M.; Ghazaly, E.; Freitas, M.O.; Mitsinga, M.; Lattanzio, L.; Nigro, C.L.; Nagano, A.; Wang, J.; Chelala, C.; Szlosarek, P.; et al. Inhibition of the Polyamine Synthesis Pathway Is Synthetically Lethal with Loss of Argininosuccinate Synthase 1. Cell Rep. 2016, 16, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochocki, J.D.; Khare, S.; Hess, M.; Ackerman, D.; Qiu, B.; Daisak, J.I.; Worth, A.J.; Lin, N.; Lee, P.; Xie, H.; et al. Arginase 2 Suppresses Renal Carcinoma Progression via Biosynthetic Cofactor Pyridoxal Phosphate Depletion and Increased Polyamine Toxicity. Cell Metab. 2018, 27, 1263–1280.e1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, F.; Sun, Y.; Xu, Z.; Niu, L.; Wang, Z.; Deng, S.; Liu, Z.; Zhou, H.; Bai, J.; Yin, Q.; et al. Excessive Polyamine Generation in Keratinocytes Promotes Self-RNA Sensing by Dendritic Cells in Psoriasis. Immunity 2020, 53, 204–216.e210. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakanishi, S.; Cleveland, J.L. Polyamine Homeostasis in Development and Disease. Med. Sci. 2021, 9, 28. https://doi.org/10.3390/medsci9020028
Nakanishi S, Cleveland JL. Polyamine Homeostasis in Development and Disease. Medical Sciences. 2021; 9(2):28. https://doi.org/10.3390/medsci9020028
Chicago/Turabian StyleNakanishi, Shima, and John L. Cleveland. 2021. "Polyamine Homeostasis in Development and Disease" Medical Sciences 9, no. 2: 28. https://doi.org/10.3390/medsci9020028
APA StyleNakanishi, S., & Cleveland, J. L. (2021). Polyamine Homeostasis in Development and Disease. Medical Sciences, 9(2), 28. https://doi.org/10.3390/medsci9020028