
Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors


Abstract
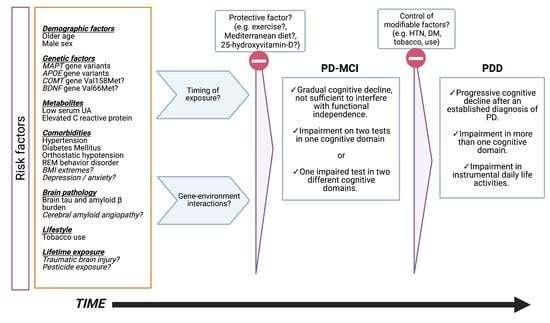
:
1. Introduction
2. Clinical profile
2.1. Cognitive Profile in Parkinson Disease
2.2. Diagnostic Criteria
2.3. Prevalence and Incidence of Cognitive Impairment in Sporadic and Genetic Forms of PD
3. Risk Factors for Cognitive Decline in Parkinson Disease
3.1. Sex
3.2. Comorbidities
3.2.1. Alzheimer Disease Pathology
3.2.2. Sleep and Mood Disorders
3.2.3. Cardiovascular Risk Factors
3.2.4. Uric Acid
3.3. Inflammation and Oxidative Stress
3.4. Environmental Risk Factors
3.4.1. Traumatic Brain Injury
3.4.2. Pesticide Exposure
3.4.3. Tobacco
3.5. Genetic Risk Factors
4. Protective Factors
4.1. Exercise
4.2. Diet
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levy, G.; Tang, M.X.; Louis, E.D.; Cote, L.J.; Alfaro, B.; Mejia, H.; Stern, Y.; Marder, K. The association of incident dementia with mortality in PD. Neurology 2002, 59, 1708–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, Y.; von Campenhausen, S.; Arend, M.; Longo, K.; Boetzel, K.; Eggert, K.; Oertel, W.H.; Dodel, R.; Barone, P. Health-related quality of life and its determinants in Parkinson’s disease: Results of an Italian cohort study. Parkinsonism Relat. Disord. 2011, 17, 265–269. [Google Scholar] [CrossRef]
- Lang, A.E.; Obeso, J.A. Time to move beyond nigrostriatal dopamine deficiency in Parkinson’s disease. Ann. Neurol. 2004, 55, 761–765. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D. Prodromal Parkinson’s Disease: The Decade Past, the Decade to Come. Mov. Disord. 2019, 34, 665–675. [Google Scholar] [CrossRef]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B.; MDS Task Force on the Definition of Parkinson’s Disease. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Darweesh, S.K.L.; Wolters, F.J.; Postuma, R.B.; Stricker, B.H.; Hofman, A.; Koudstaal, P.J.; Ikram, M.K.; Ikram, M.A. Association Between Poor Cognitive Functioning and Risk of Incident Parkinsonism: The Rotterdam Study. JAMA Neurol. 2017, 74, 1431–1438. [Google Scholar] [CrossRef]
- Schrag, A.; Anastasiou, Z.; Ambler, G.; Noyce, A.; Walters, K. Predicting diagnosis of Parkinson’s disease: A risk algorithm based on primary care presentations. Mov. Disord. 2019, 34, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Chahine, L.M.; Hawkins, K.A.; Siderowf, A.; Eberly, S.; Oakes, D.; Seibyl, J.; Stern, M.B.; Marek, K.; Jennings, D.; et al. Cognition and the course of prodromal Parkinson’s disease. Mov. Disord. 2017, 32, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Fereshtehnejad, S.M.; Yao, C.; Pelletier, A.; Montplaisir, J.Y.; Gagnon, J.F.; Postuma, R.B. Evolution of prodromal Parkinson’s disease and dementia with Lewy bodies: A prospective study. Brain 2019, 142, 2051–2067. [Google Scholar] [CrossRef]
- Darweesh, S.K.; Verlinden, V.J.; Stricker, B.H.; Hofman, A.; Koudstaal, P.J.; Ikram, M.A. Trajectories of prediagnostic functioning in Parkinson’s disease. Brain 2017, 140, 429–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougea, A.; Maraki, M.I.; Yannakoulia, M.; Stamelou, M.; Xiromerisiou, G.; Kosmidis, M.H.; Ntanasi, E.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; et al. Higher probability of prodromal Parkinson disease is related to lower cognitive performance. Neurology 2019, 92, e2261–e2272. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erro, R.; Santangelo, G.; Barone, P.; Picillo, M.; Amboni, M.; Longo, K.; Giordano, F.; Moccia, M.; Allocca, R.; Pellecchia, M.T.; et al. Do subjective memory complaints herald the onset of mild cognitive impairment in Parkinson disease? J. Geriatr. Psychiatry Neurol. 2014, 27, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, K.; Duhamel, A.; Delliaux, M.; Thomas-Anterion, C.; Destee, A.; Defebvre, L. Cognitive complaints in Parkinson’s disease: Its relationship with objective cognitive decline. J. Neurol. 2010, 257, 79–84. [Google Scholar] [CrossRef]
- Yarnall, A.J.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Coleman, S.Y.; Firbank, M.J.; Nombela, C.; Winder-Rhodes, S.; Evans, J.R.; Rowe, J.B.; et al. Characterizing mild cognitive impairment in incident Parkinson disease: The ICICLE-PD study. Neurology 2014, 82, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D.; Bronnick, K.; Williams-Gray, C.; Weintraub, D.; Marder, K.; Kulisevsky, J.; Burn, D.; Barone, P.; Pagonabarraga, J.; Allcock, L.; et al. Mild cognitive impairment in Parkinson disease: A multicenter pooled analysis. Neurology 2010, 75, 1062–1069. [Google Scholar] [CrossRef]
- Litvan, I.; Aarsland, D.; Adler, C.H.; Goldman, J.G.; Kulisevsky, J.; Mollenhauer, B.; Rodriguez-Oroz, M.C.; Troster, A.I.; Weintraub, D. MDS Task Force on mild cognitive impairment in Parkinson’s disease: Critical review of PD-MCI. Mov. Disord. 2011, 26, 1814–1824. [Google Scholar] [CrossRef] [Green Version]
- Biundo, R.; Weis, L.; Facchini, S.; Formento-Dojot, P.; Vallelunga, A.; Pilleri, M.; Antonini, A. Cognitive profiling of Parkinson disease patients with mild cognitive impairment and dementia. Parkinsonism Relat. Disord. 2014, 20, 394–399. [Google Scholar] [CrossRef]
- Hobson, P.; Meara, J. Mild cognitive impairment in Parkinson’s disease and its progression onto dementia: A 16-year outcome evaluation of the Denbighshire cohort. Int. J. Geriatr. Psychiatry 2015, 30, 1048–1055. [Google Scholar] [CrossRef]
- Kehagia, A.A.; Barker, R.A.; Robbins, T.W. Cognitive impairment in Parkinson’s disease: The dual syndrome hypothesis. Neurodegener. Dis. 2013, 11, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.N.; Cronin-Golomb, A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov. Disord. 2010, 25, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Broeders, M.; de Bie, R.M.; Velseboer, D.C.; Speelman, J.D.; Muslimovic, D.; Schmand, B. Evolution of mild cognitive impairment in Parkinson disease. Neurology 2013, 81, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, J.; Boel, J.A.; de Bie, R.M.A.; Geskus, R.B.; Schmand, B.A.; Dalrymple-Alford, J.C.; Marras, C.; Adler, C.H.; Goldman, J.G.; Troster, A.I.; et al. Mild cognitive impairment as a risk factor for Parkinson’s disease dementia. Mov. Disord. 2017, 32, 1056–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 2007, 22, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Litvan, I.; Goldman, J.G.; Troster, A.I.; Schmand, B.A.; Weintraub, D.; Petersen, R.C.; Mollenhauer, B.; Adler, C.H.; Marder, K.; Williams-Gray, C.H.; et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 2012, 27, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogland, J.; Boel, J.A.; de Bie, R.M.A.; Schmand, B.A.; Geskus, R.B.; Dalrymple-Alford, J.C.; Marras, C.; Adler, C.H.; Weintraub, D.; Junque, C.; et al. Risk of Parkinson’s disease dementia related to level I MDS PD-MCI. Mov. Disord. 2019, 34, 430–435. [Google Scholar] [CrossRef]
- Galtier, I.; Nieto, A.; Lorenzo, J.N.; Barroso, J. Mild cognitive impairment in Parkinson’s disease: Diagnosis and progression to dementia. J. Clin. Exp. Neuropsychol. 2016, 38, 40–50. [Google Scholar] [CrossRef]
- Szeto, J.Y.; Lewis, S.J. Current Treatment Options for Alzheimer’s Disease and Parkinson’s Disease Dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef]
- Skorvanek, M.; Goldman, J.G.; Jahanshahi, M.; Marras, C.; Rektorova, I.; Schmand, B.; van Duijn, E.; Goetz, C.G.; Weintraub, D.; Stebbins, G.T.; et al. Global scales for cognitive screening in Parkinson’s disease: Critique and recommendations. Mov. Disord. 2018, 33, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.G.; Holden, S.; Ouyang, B.; Bernard, B.; Goetz, C.G.; Stebbins, G.T. Diagnosing PD-MCI by MDS Task Force criteria: How many and which neuropsychological tests? Mov. Disord. 2015, 30, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogland, J.; van Wanrooij, L.L.; Boel, J.A.; Goldman, J.G.; Stebbins, G.T.; Dalrymple-Alford, J.C.; Marras, C.; Adler, C.H.; Junque, C.; Pedersen, K.F.; et al. Detecting Mild Cognitive Deficits in Parkinson’s Disease: Comparison of Neuropsychological Tests. Mov. Disord. 2018, 33, 1750–1759. [Google Scholar] [CrossRef]
- Pedersen, K.F.; Larsen, J.P.; Tysnes, O.B.; Alves, G. Natural course of mild cognitive impairment in Parkinson disease: A 5-year population-based study. Neurology 2017, 88, 767–774. [Google Scholar] [CrossRef]
- Baiano, C.; Barone, P.; Trojano, L.; Santangelo, G. Prevalence and clinical aspects of mild cognitive impairment in Parkinson’s disease: A meta-analysis. Mov. Disord. 2020, 35, 45–54. [Google Scholar] [CrossRef]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Nielsen, H.; Kragh-Sorensen, P. Risk of dementia in Parkinson’s disease: A community-based, prospective study. Neurology 2001, 56, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blazquez, C.; Schrag, A.; Rizos, A.; Chaudhuri, K.R.; Martinez-Martin, P.; Weintraub, D. Prevalence of Non-Motor Symptoms and Non-Motor Fluctuations in Parkinson’s Disease Using the MDS-NMS. Mov. Disord. Clin. Pract 2021, 8, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Geurtsen, G.J.; Hoogland, J.; Goldman, J.G.; Schmand, B.A.; Troster, A.I.; Burn, D.J.; Litvan, I.; on behalf of the MDS Study Group on the Validation of PD-MCI Criteria. Parkinson’s disease mild cognitive impairment: Application and validation of the criteria. J. Parkinsons Dis. 2014, 4, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Janvin, C.C.; Larsen, J.P.; Aarsland, D.; Hugdahl, K. Subtypes of mild cognitive impairment in Parkinson’s disease: Progression to dementia. Mov. Disord. 2006, 21, 1343–1349. [Google Scholar] [CrossRef]
- Pigott, K.; Rick, J.; Xie, S.X.; Hurtig, H.; Chen-Plotkin, A.; Duda, J.E.; Morley, J.F.; Chahine, L.M.; Dahodwala, N.; Akhtar, R.S.; et al. Longitudinal study of normal cognition in Parkinson disease. Neurology 2015, 85, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Galtier, I.; Nieto, A.; Lorenzo, J.N.; Barroso, J. Subjective cognitive decline and progression to dementia in Parkinson’s disease: A long-term follow-up study. J. Neurol. 2019, 266, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.A.; Schneider, R.B.; Saint-Hilaire, M.; Ross, G.W.; Hauser, R.A.; Lang, A.E.; Halverson, M.J.; Oakes, D.; Eberly, S.; Litvan, I.; et al. Cognitive impairment in Parkinson’s disease: Associations between subjective and objective cognitive decline in a large longitudinal study. Parkinsonism Relat. Disord. 2020, 80, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Trotta, L.; Guella, I.; Solda, G.; Sironi, F.; Tesei, S.; Canesi, M.; Pezzoli, G.; Goldwurm, S.; Duga, S.; Asselta, R. SNCA and MAPT genes: Independent and joint effects in Parkinson disease in the Italian population. Parkinsonism Relat. Disord. 2012, 18, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, Q.Y.; Zhao, Y.W.; Shu, L.; Guo, J.F.; Xu, Q.; Yan, X.X.; Tang, B.S. Effect of GBA Mutations on Phenotype of Parkinson’s Disease: A Study on Chinese Population and a Meta-Analysis. Parkinsons Dis. 2015, 2015, 916971. [Google Scholar] [CrossRef] [Green Version]
- Creese, B.; Bell, E.; Johar, I.; Francis, P.; Ballard, C.; Aarsland, D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: Review and meta-analyses. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Jesus, S.; Huertas, I.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Caceres-Redondo, M.T.; Vargas-Gonzalez, L.; Gomez-Llamas, M.; Carrillo, F.; Calderon, E.; Carballo, M.; et al. GBA Variants Influence Motor and Non-Motor Features of Parkinson’s Disease. PLoS ONE 2016, 11, e0167749. [Google Scholar] [CrossRef]
- Liu, G.; Boot, B.; Locascio, J.J.; Jansen, I.E.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; van Hilten, J.J.; et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef]
- Simuni, T.; Brumm, M.C.; Uribe, L.; Caspell-Garcia, C.; Coffey, C.S.; Siderowf, A.; Alcalay, R.N.; Trojanowski, J.Q.; Shaw, L.M.; Seibyl, J.; et al. Clinical and Dopamine Transporter Imaging Characteristics of Leucine Rich Repeat Kinase 2 (LRRK2) and Glucosylceramidase Beta (GBA) Parkinson’s Disease Participants in the Parkinson’s Progression Markers Initiative: A Cross-Sectional Study. Mov. Disord. 2020, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Mirelman, A.; Saunders-Pullman, R.; Tang, M.X.; Mejia Santana, H.; Raymond, D.; Roos, E.; Orbe-Reilly, M.; Gurevich, T.; Bar Shira, A.; et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov. Disord. 2013, 28, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Piredda, R.; Desmarais, P.; Masellis, M.; Gasca-Salas, C. Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: A systematic review. Eur. J. Neurol. 2020, 27, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef]
- Grunewald, A.; Kasten, M.; Ziegler, A.; Klein, C. Next-generation phenotyping using the parkin example: Time to catch up with genetics. JAMA Neurol. 2013, 70, 1186–1191. [Google Scholar] [CrossRef]
- Szewczyk-Krolikowski, K.; Tomlinson, P.; Nithi, K.; Wade-Martins, R.; Talbot, K.; Ben-Shlomo, Y.; Hu, M.T. The influence of age and gender on motor and non-motor features of early Parkinson’s disease: Initial findings from the Oxford Parkinson Disease Center (OPDC) discovery cohort. Parkinsonism Relat. Disord. 2014, 20, 99–105. [Google Scholar] [CrossRef]
- Augustine, E.F.; Perez, A.; Dhall, R.; Umeh, C.C.; Videnovic, A.; Cambi, F.; Wills, A.M.; Elm, J.J.; Zweig, R.M.; Shulman, L.M.; et al. Sex Differences in Clinical Features of Early, Treated Parkinson’s Disease. PLoS ONE 2015, 10, e0133002. [Google Scholar] [CrossRef]
- Gao, L.; Nie, K.; Tang, H.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Feng, S.; Zhu, R.; Duan, Z.; et al. Sex differences in cognition among Chinese people with Parkinson’s disease. J. Clin. Neurosci. 2015, 22, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Umbach, D.M.; Peddada, S.D.; Xu, Z.; Troster, A.I.; Huang, X.; Chen, H. Potential sex differences in nonmotor symptoms in early drug-naive Parkinson disease. Neurology 2015, 84, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Munro, C.A.; Winicki, J.M.; Schretlen, D.J.; Gower, E.W.; Turano, K.A.; Munoz, B.; Keay, L.; Bandeen-Roche, K.; West, S.K. Sex differences in cognition in healthy elderly individuals. Neuropsychol. Dev. Cogn. B Aging Neuropsychol. Cogn. 2012, 19, 759–768. [Google Scholar] [CrossRef]
- Sundermann, E.E.; Maki, P.M.; Rubin, L.H.; Lipton, R.B.; Landau, S.; Biegon, A.; Alzheimer’s Disease Neuroimaging Initiative. Female advantage in verbal memory: Evidence of sex-specific cognitive reserve. Neurology 2016, 87, 1916–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, T.; Liu, C.; Ginghina, C.; Cain, K.C.; Auinger, P.; Cholerton, B.; Shi, M.; Zhang, J.; Parkinson Study Group DATATOP Investigators. Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am. J. Pathol. 2014, 184, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compta, Y.; Valente, T.; Saura, J.; Segura, B.; Iranzo, A.; Serradell, M.; Junque, C.; Tolosa, E.; Valldeoriola, F.; Munoz, E.; et al. Correlates of cerebrospinal fluid levels of oligomeric- and total-alpha-synuclein in premotor, motor and dementia stages of Parkinson’s disease. J. Neurol. 2015, 262, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Hall, S.; Ohrfelt, A.; Zetterberg, H.; Blennow, K.; Minthon, L.; Nagga, K.; Londos, E.; Varghese, S.; Majbour, N.K.; et al. Levels of cerebrospinal fluid alpha-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, D.G.; Hurtig, H.I.; Irwin, D.J. Pathological Influences on Clinical Heterogeneity in Lewy Body Diseases. Mov. Disord. 2020, 35, 5–19. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.; Malek, N.; Grosset, K.; Cullen, B.; Gentleman, S.; Grosset, D.G. Neuropathology of dementia in patients with Parkinson’s disease: A systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, B.; Trenkwalder, C.; von Ahsen, N.; Bibl, M.; Steinacker, P.; Brechlin, P.; Schindehuette, J.; Poser, S.; Wiltfang, J.; Otto, M. Beta-amlyoid 1-42 and tau-protein in cerebrospinal fluid of patients with Parkinson’s disease dementia. Dement. Geriatr. Cogn. Disord. 2006, 22, 200–208. [Google Scholar] [CrossRef]
- Montine, T.J.; Shi, M.; Quinn, J.F.; Peskind, E.R.; Craft, S.; Ginghina, C.; Chung, K.A.; Kim, H.; Galasko, D.R.; Jankovic, J.; et al. CSF Abeta(42) and tau in Parkinson’s disease with cognitive impairment. Mov. Disord. 2010, 25, 2682–2685. [Google Scholar] [CrossRef] [Green Version]
- Compta, Y.; Pereira, J.B.; Rios, J.; Ibarretxe-Bilbao, N.; Junque, C.; Bargallo, N.; Camara, A.; Buongiorno, M.; Fernandez, M.; Pont-Sunyer, C.; et al. Combined dementia-risk biomarkers in Parkinson’s disease: A prospective longitudinal study. Parkinsonism Relat. Disord. 2013, 19, 717–724. [Google Scholar] [CrossRef]
- Vranova, H.P.; Henykova, E.; Kaiserova, M.; Mensikova, K.; Vastik, M.; Mares, J.; Hlustik, P.; Zapletalova, J.; Strnad, M.; Stejskal, D.; et al. Tau protein, beta-amyloid(1)(-)(4)(2) and clusterin CSF levels in the differential diagnosis of Parkinsonian syndrome with dementia. J. Neurol. Sci. 2014, 343, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Compta, Y.; Marti, M.J.; Ibarretxe-Bilbao, N.; Junque, C.; Valldeoriola, F.; Munoz, E.; Ezquerra, M.; Rios, J.; Tolosa, E. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov. Disord. 2009, 24, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.K.; Alves, G.; Hwang, K.S.; Babakchanian, S.; Bronnick, K.S.; Chou, Y.Y.; Dalaker, T.O.; Kurz, M.W.; Larsen, J.P.; Somme, J.H.; et al. Cerebrospinal fluid Abeta levels correlate with structural brain changes in Parkinson’s disease. Mov. Disord. 2013, 28, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.Y.; Zuo, L.J.; Wang, F.; Chen, Z.J.; Hu, Y.; Wang, Y.J.; Wang, X.M.; Zhang, W. Potential biomarkers relating pathological proteins, neuroinflammatory factors and free radicals in PD patients with cognitive impairment: A cross-sectional study. BMC Neurol. 2014, 14, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtig, H.I.; Trojanowski, J.Q.; Galvin, J.; Ewbank, D.; Schmidt, M.L.; Lee, V.M.; Clark, C.M.; Glosser, G.; Stern, M.B.; Gollomp, S.M.; et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54, 1916–1921. [Google Scholar] [CrossRef]
- Kovari, E.; Gold, G.; Herrmann, F.R.; Canuto, A.; Hof, P.R.; Bouras, C.; Giannakopoulos, P. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 2003, 106, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Perry, R.; Brown, A.; Larsen, J.P.; Ballard, C. Neuropathology of dementia in Parkinson’s disease: A prospective, community-based study. Ann. Neurol. 2005, 58, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Seppi, K.; Wenning, G.K.; Poewe, W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J. Neural Transm. 2002, 109, 329–339. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Cairns, N.J.; Campbell, M.C.; Willis, A.W.; Racette, B.A.; Tabbal, S.D.; Perlmutter, J.S. Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Arch. Neurol. 2012, 69, 1326–1331. [Google Scholar] [CrossRef] [Green Version]
- Compta, Y.; Parkkinen, L.; O’Sullivan, S.S.; Vandrovcova, J.; Holton, J.L.; Collins, C.; Lashley, T.; Kallis, C.; Williams, D.R.; de Silva, R.; et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 2011, 134, 1493–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, D.R.; Whitfield, D.; Johnson, M.; Attems, J.; O’Brien, J.T.; Aarsland, D.; Lai, M.K.; Lee, J.H.; Chen, C.; Ballard, C.; et al. Regional Multiple Pathology Scores Are Associated with Cognitive Decline in Lewy Body Dementias. Brain Pathol. 2015, 25, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, J.; Herrmann, F.R.; Burkhard, P.R.; Bouras, C.; Kovari, E. Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 864–868, discussion 864. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Cerebral amyloid angiopathy in Lewy body disease. J. Neural. Transm. 2008, 115, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Ling, H.; Lashley, T.; Foley, J.A.; Strand, C.; Eid, T.M.; Holton, J.L.; Warner, T.T. Novel clinicopathological characteristics differentiate dementia with Lewy bodies from Parkinson’s disease dementia. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Herman, A.M.; Khandelwal, P.J.; Stanczyk, B.B.; Rebeck, G.W.; Moussa, C.E. beta-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011, 1386, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; White, M.T.; Toledo, J.B.; Xie, S.X.; Robinson, J.L.; Van Deerlin, V.; Lee, V.M.; Leverenz, J.B.; Montine, T.J.; Duda, J.E.; et al. Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 2012, 72, 587–598. [Google Scholar] [CrossRef]
- Chahine, L.M.; Xie, S.X.; Simuni, T.; Tran, B.; Postuma, R.; Amara, A.; Oertel, W.H.; Iranzo, A.; Scordia, C.; Fullard, M.; et al. Longitudinal changes in cognition in early Parkinson’s disease patients with REM sleep behavior disorder. Parkinsonism Relat. Disord. 2016, 27, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Forbes, E.; Tropea, T.F.; Mantri, S.; Xie, S.X.; Morley, J.F. Modifiable comorbidities associated with cognitive decline in Parkinson’s Disease. Mov. Disord. Clin. Pract. 2020. [Google Scholar] [CrossRef]
- Postuma, R.B.; Bertrand, J.A.; Montplaisir, J.; Desjardins, C.; Vendette, M.; Rios Romenets, S.; Panisset, M.; Gagnon, J.F. Rapid eye movement sleep behavior disorder and risk of dementia in Parkinson’s disease: A prospective study. Mov. Disord. 2012, 27, 720–726. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, W.; Liu, F.T.; Li, J.Q.; Cao, X.P.; Tan, L.; Wang, J.; Yu, J.T. Modifiable risk factors for cognitive impairment in Parkinson’s disease: A systematic review and meta-analysis of prospective cohort studies. Mov. Disord. 2019, 34, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Laatikainen, T.; Winblad, B.; Soininen, H.; Tuomilehto, J. Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol. 2006, 5, 735–741. [Google Scholar] [CrossRef]
- Chahine, L.M.; Dos Santos, C.; Fullard, M.; Scordia, C.; Weintraub, D.; Erus, G.; Rosenthal, L.; Davatzikos, C.; McMillan, C.T. Modifiable vascular risk factors, white matter disease and cognition in early Parkinson’s disease. Eur. J. Neurol. 2019, 26, 246-e18. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Nabizadeh, N.; Caunca, M.; Cheung, Y.K.; Rundek, T.; Elkind, M.S.; DeCarli, C.; Sacco, R.L.; Stern, Y.; Wright, C.B. Cognitive correlates of white matter lesion load and brain atrophy: The Northern Manhattan Study. Neurology 2015, 85, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Erten-Lyons, D.; Woltjer, R.; Kaye, J.; Mattek, N.; Dodge, H.H.; Green, S.; Tran, H.; Howieson, D.B.; Wild, K.; Silbert, L.C. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 2013, 81, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, B.; Zimmermann, J.; Sixel-Doring, F.; Focke, N.K.; Wicke, T.; Ebentheuer, J.; Schaumburg, M.; Lang, E.; Friede, T.; Trenkwalder, C.; et al. Baseline predictors for progression 4 years after Parkinson’s disease diagnosis in the De Novo Parkinson Cohort (DeNoPa). Mov. Disord. 2019, 34, 67–77. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, E.S.; Lee, J.H.; Moon, J.S.; Oh, J.E.; Shin, J.W.; Lee, K.J.; Baek, I.C.; Jeong, S.H.; Song, H.J.; et al. Relationship between changes of body mass index (BMI) and cognitive decline in Parkinson’s disease (PD). Arch. Gerontol. Geriatr. 2012, 55, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.S.; Chung, S.J.; Lee, P.H.; Sohn, Y.H.; Kang, S.Y. The Influence of Body Mass Index at Diagnosis on Cognitive Decline in Parkinson’s Disease. J. Clin. Neurol. 2019, 15, 517–526. [Google Scholar] [CrossRef]
- Anang, J.B.; Gagnon, J.F.; Bertrand, J.A.; Romenets, S.R.; Latreille, V.; Panisset, M.; Montplaisir, J.; Postuma, R.B. Predictors of dementia in Parkinson disease: A prospective cohort study. Neurology 2014, 83, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.D.; Udow, S.J.; Espay, A.J.; Merola, A.; Camicioli, R.; Lang, A.E.; Masellis, M. Orthostatic hypotension and dementia incidence: Links and implications. Neuropsychiatr. Dis. Treat. 2019, 15, 2181–2194. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, S.; Gonzalez, J.; Mash, D.C. The effect of ischemic cerebrovascular disease on the clinical characteristics of Parkinson’s disease. A post-mortem study. Eur. J. Neurol. 2006, 13, 96–97. [Google Scholar] [CrossRef]
- Ghebremedhin, E.; Rosenberger, A.; Rub, U.; Vuksic, M.; Berhe, T.; Bickeboller, H.; de Vos, R.A.; Thal, D.R.; Deller, T. Inverse relationship between cerebrovascular lesions and severity of lewy body pathology in patients with lewy body diseases. J. Neuropathol. Exp. Neurol. 2010, 69, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Moran, L.B.; Graeber, M.B. Towards a pathway definition of Parkinson’s disease: A complex disorder with links to cancer, diabetes and inflammation. Neurogenetics 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.; Farina, C. Shared molecular and functional frameworks among five complex human disorders: A comparative study on interactomes linked to susceptibility genes. PLoS ONE 2011, 6, e18660. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Parkinsons Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.A.; Potashkin, J.A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 2013, 19, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.; Plastino, M.; Cristiano, D.; Colica, C.; Ermio, C.; De Bartolo, M.; Mungari, P.; Fonte, G.; Consoli, D.; Consoli, A.; et al. Dementia is associated with insulin resistance in patients with Parkinson’s disease. J. Neurol. Sci. 2012, 315, 39–43. [Google Scholar] [CrossRef]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef]
- Chung, S.J.; Jeon, S.; Yoo, H.S.; Kim, G.; Oh, J.S.; Kim, J.S.; Evans, A.C.; Sohn, Y.H.; Lee, P.H. Detrimental effect of type 2 diabetes mellitus in a large case series of Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 64, 54–59. [Google Scholar] [CrossRef]
- Shen, C.; Guo, Y.; Luo, W.; Lin, C.; Ding, M. Serum urate and the risk of Parkinson’s disease: Results from a meta-analysis. Can. J. Neurol. Sci. 2013, 40, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Weisskopf, M.G.; O’Reilly, E.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Plasma urate and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 166, 561–567. [Google Scholar] [CrossRef]
- Jain, S.; Ton, T.G.; Boudreau, R.M.; Yang, M.; Thacker, E.L.; Studenski, S.; Longstreth, W.T., Jr.; Strotmeyer, E.S.; Newman, A.B. The risk of Parkinson disease associated with urate in a community-based cohort of older adults. Neuroepidemiology 2011, 36, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Quinn, T.J.; Hewitt, J.; Fan, Y.; Dawson, J. Serum uric acid level and association with cognitive impairment and dementia: Systematic review and meta-analysis. Age 2016, 38, 16. [Google Scholar] [CrossRef] [Green Version]
- Maetzler, W.; Stapf, A.K.; Schulte, C.; Hauser, A.K.; Lerche, S.; Wurster, I.; Schleicher, E.; Melms, A.; Berg, D. Serum and cerebrospinal fluid uric acid levels in lewy body disorders: Associations with disease occurrence and amyloid-beta pathway. J. Alzheimer’s Dis. 2011, 27, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Annanmaki, T.; Pessala-Driver, A.; Hokkanen, L.; Murros, K. Uric acid associates with cognition in Parkinson’s disease. Parkinsonism Relat. Disord. 2008, 14, 576–578. [Google Scholar] [CrossRef]
- Moccia, M.; Picillo, M.; Erro, R.; Vitale, C.; Longo, K.; Amboni, M.; Santangelo, G.; Spina, E.; De Rosa, A.; De Michele, G.; et al. Is serum uric acid related to non-motor symptoms in de-novo Parkinson’s disease patients? Parkinsonism Relat. Disord. 2014, 20, 772–775. [Google Scholar] [CrossRef]
- Moccia, M.; Picillo, M.; Erro, R.; Vitale, C.; Longo, K.; Amboni, M.; Santangelo, G.; Palladino, R.; Capo, G.; Orefice, G.; et al. Presence and progression of non-motor symptoms in relation to uric acid in de novo Parkinson’s disease. Eur. J. Neurol. 2015, 22, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Aman, Y.; Ahmed, I.; Chetelat, G.; Landeau, B.; Ray Chaudhuri, K.; Brooks, D.J.; Edison, P. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimer’s Dement. 2015, 11, 608–621.e7. [Google Scholar] [CrossRef]
- Lindqvist, D.; Hall, S.; Surova, Y.; Nielsen, H.M.; Janelidze, S.; Brundin, L.; Hansson, O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease--associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 2013, 136, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garabadu, D.; Agrawal, N.; Sharma, A.; Sharma, S. Mitochondrial metabolism: A common link between neuroinflammation and neurodegeneration. Behav. Pharmacol. 2019, 30, 642–652. [Google Scholar] [CrossRef]
- Jeon, Y.M.; Kwon, Y.; Jo, M.; Lee, S.; Kim, S.; Kim, H.J. The Role of Glial Mitochondria in alpha-Synuclein Toxicity. Front. Cell Dev. Biol. 2020, 8, 548283. [Google Scholar] [CrossRef] [PubMed]
- Gatt, A.P.; Duncan, O.F.; Attems, J.; Francis, P.T.; Ballard, C.G.; Bateman, J.M. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord. 2016, 31, 352–359. [Google Scholar] [CrossRef]
- Toklu, H.Z.; Tumer, N. Oxidative Stress, Brain Edema, Blood-Brain Barrier Permeability, and Autonomic Dysfunction from Traumatic Brain Injury. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; Frontiers in Neuroengineering: Boca Raton, FL, USA, 2015. [Google Scholar]
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association of Traumatic Brain Injury With Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol. 2016, 73, 1062–1069. [Google Scholar] [CrossRef]
- Goldman, S.M.; Tanner, C.M.; Oakes, D.; Bhudhikanok, G.S.; Gupta, A.; Langston, J.W. Head injury and Parkinson’s disease risk in twins. Ann. Neurol. 2006, 60, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Baldereschi, M.; Di Carlo, A.; Vanni, P.; Ghetti, A.; Carbonin, P.; Amaducci, L.; Inzitari, D.; for the Italian Longitudinal Study on Aging Working Group. Lifestyle-related risk factors for Parkinson’s disease: A population-based study. Acta Neurol. Scand. 2003, 108, 239–244. [Google Scholar] [CrossRef]
- Marras, C.; Hincapie, C.A.; Kristman, V.L.; Cancelliere, C.; Soklaridis, S.; Li, A.; Borg, J.; af Geijerstam, J.L.; Cassidy, J.D. Systematic review of the risk of Parkinson’s disease after mild traumatic brain injury: Results of the International Collaboration on Mild Traumatic Brain Injury Prognosis. Arch. Phys. Med. Rehabil. 2014, 95, S238–S244. [Google Scholar] [CrossRef]
- Krauss, J.K. Movement disorders secondary to craniocerebral trauma. Handb. Clin. Neurol. 2015, 128, 475–496. [Google Scholar] [CrossRef]
- Schiehser, D.M.; Filoteo, J.V.; Litvan, I.; Pirogovsky-Turk, E.; Lessig, S.L.; Song, D.S. Cognitive functioning in individuals with Parkinson’s disease and traumatic brain injury: A longitudinal study. Parkinsonism Relat. Disord. 2016, 30, 58–61. [Google Scholar] [CrossRef]
- Tanner, C.M.; Ross, G.W.; Jewell, S.A.; Hauser, R.A.; Jankovic, J.; Factor, S.A.; Bressman, S.; Deligtisch, A.; Marras, C.; Lyons, K.E.; et al. Occupation and risk of parkinsonism: A multicenter case-control study. Arch. Neurol. 2009, 66, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Brown, E.; Meng, C.; Goldman, S.; Tanner, C. Pesticide Exposure in PD with and without Genetic Risk Variants. In Proceedings of the Parkinson Disease and Movement Disorders Society Virtual Congress, International Parkinson and Movement Disorder Society, Milwaukee, WI, USA, 12–16 September 2020. [Google Scholar]
- Hernan, M.A.; Zhang, S.M.; Rueda-deCastro, A.M.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Cigarette smoking and the incidence of Parkinson’s disease in two prospective studies. Ann. Neurol. 2001, 50, 780–786. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.; Guo, X.; Mailman, R.B.; Park, Y.; Kamel, F.; Umbach, D.M.; Xu, Q.; Hollenbeck, A.; Schatzkin, A.; et al. Smoking duration, intensity, and risk of Parkinson disease. Neurology 2010, 74, 878–884. [Google Scholar] [CrossRef]
- Allam, M.F.; Campbell, M.J.; Hofman, A.; Del Castillo, A.S.; Fernandez-Crehuet Navajas, R. Smoking and Parkinson’s disease: Systematic review of prospective studies. Mov. Disord. 2004, 19, 614–621. [Google Scholar] [CrossRef]
- Paul, K.C.; Chuang, Y.H.; Shih, I.F.; Keener, A.; Bordelon, Y.; Bronstein, J.M.; Ritz, B. The association between lifestyle factors and Parkinson’s disease progression and mortality. Mov. Disord. 2019, 34, 58–66. [Google Scholar] [CrossRef]
- Doiron, M.; Dupre, N.; Langlois, M.; Provencher, P.; Simard, M. Smoking history is associated to cognitive impairment in Parkinson’s disease. Aging Ment. Health 2017, 21, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Luca, A.; Baschi, R.; Cicero, C.E.; Mostile, G.; Davi, M.; La Bianca, G.; Restivo, V.; Zappia, M.; Monastero, R. Vascular risk factors, white matter lesions and cognitive impairment in Parkinson’s disease: The PACOS longitudinal study. J. Neurol. 2021, 268, 549–558. [Google Scholar] [CrossRef]
- Nombela, C.; Rowe, J.B.; Winder-Rhodes, S.E.; Hampshire, A.; Owen, A.M.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Yarnall, A.J.; Firbank, M.J.; et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain 2014, 137, 2743–2758. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Rausch, R.; Creek, M.M.; Sinsheimer, J.S.; Bronstein, J.M.; Bordelon, Y.; Ritz, B. APOE, MAPT, and COMT and Parkinson’s Disease Susceptibility and Cognitive Symptom Progression. J. Parkinsons Dis. 2016, 6, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Williams-Gray, C.H.; Goris, A.; Saiki, M.; Foltynie, T.; Compston, D.A.; Sawcer, S.J.; Barker, R.A. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J. Neurol. 2009, 256, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Mason, S.L.; Evans, J.R.; Foltynie, T.; Brayne, C.; Robbins, T.W.; Barker, R.A. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1258–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto-Salvia, N.; Clarimon, J.; Pagonabarraga, J.; Pascual-Sedano, B.; Campolongo, A.; Combarros, O.; Mateo, J.I.; Regana, D.; Martinez-Corral, M.; Marquie, M.; et al. Dementia risk in Parkinson disease: Disentangling the role of MAPT haplotypes. Arch. Neurol. 2011, 68, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.F.; Xie, S.X.; Hurtig, H.I.; Stern, M.B.; Colcher, A.; Horn, S.; Dahodwala, N.; Duda, J.E.; Weintraub, D.; Chen-Plotkin, A.S.; et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov. Disord. 2012, 27, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Hurtig, H.I.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Rhodes, S.L.; Factor, S.A.; et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014, 71, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.M.; Williams-Gray, C.H. The Genetic Basis of Cognitive Impairment and Dementia in Parkinson’s Disease. Front. Psychiatry 2016, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lipska, B.K.; Halim, N.; Ma, Q.D.; Matsumoto, M.; Melhem, S.; Kolachana, B.S.; Hyde, T.M.; Herman, M.M.; Apud, J.; et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): Effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 2004, 75, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Barnett, J.H.; Jones, P.B.; Robbins, T.W.; Muller, U. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: A meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol. Psychiatry 2007, 12, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Foltynie, T.; Goldberg, T.E.; Lewis, S.G.; Blackwell, A.D.; Kolachana, B.S.; Weinberger, D.R.; Robbins, T.W.; Barker, R.A. Planning ability in Parkinson’s disease is influenced by the COMT val158met polymorphism. Mov. Disord. 2004, 19, 885–891. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Evans, J.R.; Goris, A.; Foltynie, T.; Ban, M.; Robbins, T.W.; Brayne, C.; Kolachana, B.S.; Weinberger, D.R.; Sawcer, S.J.; et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 2009, 132, 2958–2969. [Google Scholar] [CrossRef] [Green Version]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y.; Patel, P.D.; Sant, G.; Meng, C.X.; Teng, K.K.; Hempstead, B.L.; Lee, F.S. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J. Neurosci. 2004, 24, 4401–4411. [Google Scholar] [CrossRef]
- Guerini, F.R.; Beghi, E.; Riboldazzi, G.; Zangaglia, R.; Pianezzola, C.; Bono, G.; Casali, C.; Di Lorenzo, C.; Agliardi, C.; Nappi, G.; et al. BDNF Val66Met polymorphism is associated with cognitive impairment in Italian patients with Parkinson’s disease. Eur. J. Neurol. 2009, 16, 1240–1245. [Google Scholar] [CrossRef]
- Bialecka, M.; Kurzawski, M.; Roszmann, A.; Robowski, P.; Sitek, E.J.; Honczarenko, K.; Mak, M.; Deptula-Jarosz, M.; Golab-Janowska, M.; Drozdzik, M.; et al. BDNF G196A (Val66Met) polymorphism associated with cognitive impairment in Parkinson’s disease. Neurosci. Lett. 2014, 561, 86–90. [Google Scholar] [CrossRef]
- Teh, C.A.; Lee, T.S.; Kuchibhatla, M.; Ashley-Koch, A.; Macfall, J.; Krishnan, R.; Beyer, J. Bipolar disorder, brain-derived neurotrophic factor (BDNF) Val66Met polymorphism and brain morphology. PLoS ONE 2012, 7, e38469. [Google Scholar] [CrossRef] [Green Version]
- Karakasis, C.; Kalinderi, K.; Katsarou, Z.; Fidani, L.; Bostantjopoulou, S. Association of brain-derived neurotrophic factor (BDNF) Val66Met polymorphism with Parkinson’s disease in a Greek population. J. Clin. Neurosci. 2011, 18, 1744–1745. [Google Scholar] [CrossRef]
- Svetel, M.; Pekmezovic, T.; Markovic, V.; Novakovic, I.; Dobricic, V.; Djuric, G.; Stefanova, E.; Kostic, V. No association between brain-derived neurotrophic factor G196A polymorphism and clinical features of Parkinson’s disease. Eur. Neurol. 2013, 70, 257–262. [Google Scholar] [CrossRef]
- Van der Kolk, N.M.; Speelman, A.D.; van Nimwegen, M.; Kessels, R.P.; IntHout, J.; Hakobjan, M.; Munneke, M.; Bloem, B.R.; van de Warrenburg, B.P. BDNF polymorphism associates with decline in set shifting in Parkinson’s disease. Neurobiol. Aging 2015, 36, 1605.e1–1605.e6. [Google Scholar] [CrossRef]
- Smith, P.J.; Blumenthal, J.A.; Hoffman, B.M.; Cooper, H.; Strauman, T.A.; Welsh-Bohmer, K.; Browndyke, J.N.; Sherwood, A. Aerobic exercise and neurocognitive performance: A meta-analytic review of randomized controlled trials. Psychosom. Med. 2010, 72, 239–252. [Google Scholar] [CrossRef]
- Tanaka, K.; Quadros, A.C., Jr.; Santos, R.F.; Stella, F.; Gobbi, L.T.; Gobbi, S. Benefits of physical exercise on executive functions in older people with Parkinson’s disease. Brain Cogn. 2009, 69, 435–441. [Google Scholar] [CrossRef]
- Picelli, A.; Varalta, V.; Melotti, C.; Zatezalo, V.; Fonte, C.; Amato, S.; Saltuari, L.; Santamato, A.; Fiore, P.; Smania, N. Effects of treadmill training on cognitive and motor features of patients with mild to moderate Parkinson’s disease: A pilot, single-blind, randomized controlled trial. Funct. Neurol. 2016, 31, 25–31. [Google Scholar] [CrossRef]
- David, F.J.; Robichaud, J.A.; Leurgans, S.E.; Poon, C.; Kohrt, W.M.; Goldman, J.G.; Comella, C.L.; Vaillancourt, D.E.; Corcos, D.M. Exercise improves cognition in Parkinson’s disease: The PRET-PD randomized, clinical trial. Mov. Disord. 2015, 30, 1657–1663. [Google Scholar] [CrossRef] [Green Version]
- Cruise, K.E.; Bucks, R.S.; Loftus, A.M.; Newton, R.U.; Pegoraro, R.; Thomas, M.G. Exercise and Parkinson’s: Benefits for cognition and quality of life. Acta Neurol. Scand. 2011, 123, 13–19. [Google Scholar] [CrossRef]
- Aguilo, A.; Tauler, P.; Fuentespina, E.; Tur, J.A.; Cordova, A.; Pons, A. Antioxidant response to oxidative stress induced by exhaustive exercise. Physiol. Behav. 2005, 84, 1–7. [Google Scholar] [CrossRef]
- Peijie, C.; Hongwu, L.; Fengpeng, X.; Jie, R.; Jie, Z. Heavy load exercise induced dysfunction of immunity and neuroendocrine responses in rats. Life Sci. 2003, 72, 2255–2262. [Google Scholar] [CrossRef]
- Rosa, E.F.; Takahashi, S.; Aboulafia, J.; Nouailhetas, V.L.; Oliveira, M.G. Oxidative stress induced by intense and exhaustive exercise impairs murine cognitive function. J. Neurophysiol. 2007, 98, 1820–1826. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Luchsinger, J.A.; Stern, Y.; Scarmeas, N. Mediterranean diet, inflammatory and metabolic biomarkers, and risk of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 22, 483–492. [Google Scholar] [CrossRef] [Green Version]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr. Nutr. Rep. 2019, 8, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Paknahad, Z.; Sheklabadi, E.; Derakhshan, Y.; Bagherniya, M.; Chitsaz, A. The effect of the Mediterranean diet on cognitive function in patients with Parkinson’s disease: A randomized clinical controlled trial. Complement. Ther. Med. 2020, 50, 102366. [Google Scholar] [CrossRef] [PubMed]
- Caprara, G. Mediterranean-Type Dietary Pattern and Physical Activity: The Winning Combination to Counteract the Rising Burden of Non-Communicable Diseases (NCDs). Nutrients 2021, 13, 429. [Google Scholar] [CrossRef]
- Luo, X.; Ou, R.; Dutta, R.; Tian, Y.; Xiong, H.; Shang, H. Association Between Serum Vitamin D Levels and Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2018, 9, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, I.; Griebler, U.; Kien, C.; Auer, S.; Klerings, I.; Hammer, R.; Holzer, P.; Gartlehner, G. Vitamin D deficiency as a risk factor for dementia: A systematic review and meta-analysis. BMC Geriatr. 2017, 17, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barichella, M.; Cereda, E.; Iorio, L.; Pinelli, G.; Ferri, V.; Cassani, E.; Bolliri, C.; Caronni, S.; Pusani, C.; Schiaffino, M.G.; et al. Clinical correlates of serum 25-hydroxyvitamin D in Parkinson’s disease. Nutr. Neurosci. 2020, 1–9. [Google Scholar] [CrossRef]
- Peterson, A.L.; Murchison, C.; Zabetian, C.; Leverenz, J.B.; Watson, G.S.; Montine, T.; Carney, N.; Bowman, G.L.; Edwards, K.; Quinn, J.F. Memory, mood, and vitamin D in persons with Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Sleeman, I.; Aspray, T.; Lawson, R.; Coleman, S.; Duncan, G.; Khoo, T.K.; Schoenmakers, I.; Rochester, L.; Burn, D.; Yarnall, A. The Role of Vitamin D in Disease Progression in Early Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Gelber, R.P.; Petrovitch, H.; Masaki, K.H.; Ross, G.W.; White, L.R. Coffee intake in midlife and risk of dementia and its neuropathologic correlates. J. Alzheimer’s Dis. 2011, 23, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hebert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernan, M.A.; Takkouche, B.; Caamano-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Cho, B.H.; Choi, S.M.; Kim, J.T.; Kim, B.C. Association of coffee consumption and non-motor symptoms in drug-naive, early-stage Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 50, 42–47. [Google Scholar] [CrossRef]
- Cho, B.H.; Choi, S.M.; Kim, B.C. Gender-dependent effect of coffee consumption on tremor severity in de novo Parkinson’s disease. BMC Neurol. 2019, 19, 194. [Google Scholar] [CrossRef]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as symptomatic treatment for Parkinson disease (Cafe-PD): A randomized trial. Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef]


{kind=link}
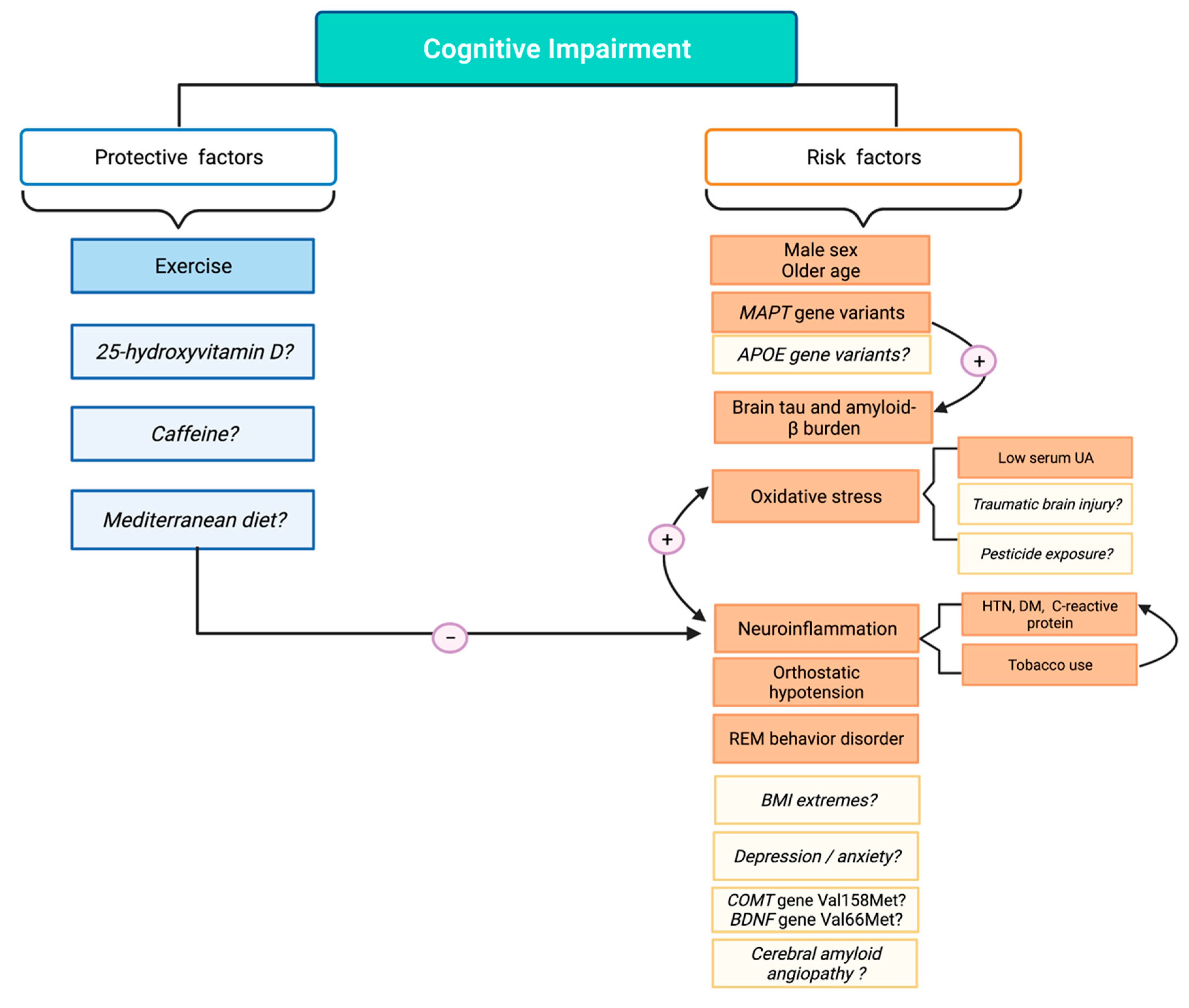{kind=link}
| Attention/ Working Memory | Trail Making Test-Part A |
|---|---|
| Symbol Digit Modalities Test | |
| Executive function | Trail Making Test-Part B |
| Clock Drawing Test | |
| Visuospatial functioning | Judgment of Line Orientation |
| Intersecting pentagons | |
| Language | Boston Naming Test |
| Animal naming | |
| Memory | Free and Cued Selective Reminding Test |
| Figural Memory |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Latapi, P.; Bayram, E.; Litvan, I.; Marras, C. Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors. Behav. Sci. 2021, 11, 74. https://doi.org/10.3390/bs11050074
Gonzalez-Latapi P, Bayram E, Litvan I, Marras C. Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors. Behavioral Sciences. 2021; 11(5):74. https://doi.org/10.3390/bs11050074
Chicago/Turabian StyleGonzalez-Latapi, Paulina, Ece Bayram, Irene Litvan, and Connie Marras. 2021. "Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors" Behavioral Sciences 11, no. 5: 74. https://doi.org/10.3390/bs11050074
APA StyleGonzalez-Latapi, P., Bayram, E., Litvan, I., & Marras, C. (2021). Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors. Behavioral Sciences, 11(5), 74. https://doi.org/10.3390/bs11050074