Copy Number Variation: Methods and Clinical Applications

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
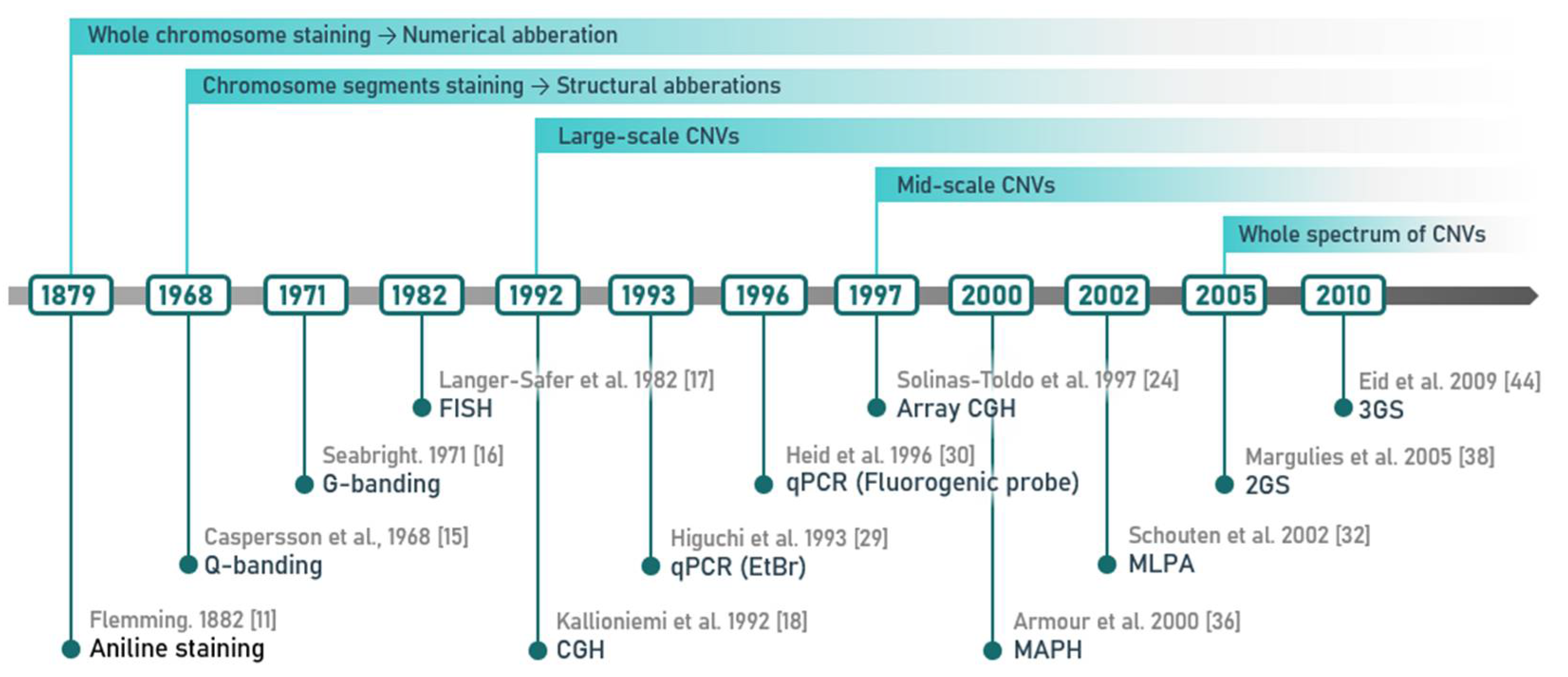
2. Methods of CNV Detection
2.1. Cytogenetic Techniques and Their Most Common Modifications
2.2. Methods of Molecular Biology
2.3. Techniques Possibly Affected by the Presence of Undetected CNVs
3. Potential Biomedical Applications of CNV Detection
4. Clinical Interpretation of CNVs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017, 58, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Gu, X.; Wang, G.; Huang, Y.; Ju, S.; Huang, J.; Wang, X. Copy number variations primed lncRNAs deregulation contribute to poor prognosis in colorectal cancer. Aging 2019, 11, 6089–6108. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.; Krishnan, P.; Cass, C.E.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Breast cancer associated germline structural variants harboring small noncoding RNAs impact post-transcriptional gene regulation. Sci. Rep. 2018, 8, 7529. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Bennett, P.M. Genome plasticity: Insertion sequence elements, transposons and integrons, and DNA rearrangement. Methods Mol. Biol. 2004, 266, 71–113. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Flemming, W. Zellsubstanz, Kern und Zelltheilung; FCW Vogel: Leipzig, Germany, 1882. [Google Scholar]
- Tice, S.C. A new sex-linked character in drosophila. Biol. Bull. 1914, 26, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Tjio, J.H.; Levan, A. The chromosome number of man. Hereditas 2010, 42, 1–6. [Google Scholar] [CrossRef]
- Moorhead, P.S.; Nowell, P.C.; Mellman, W.J.; Battips, D.M.; Hungerford, D.A. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp. Cell Res. 1960, 20, 613–616. [Google Scholar] [CrossRef]
- Caspersson, T.; Farber, S.; Foley, G.E.; Kudynowski, J.; Modest, E.J.; Simonsson, E.; Wagh, U.; Zech, L. Chemical differentiation along metaphase chromosomes. Exp. Cell Res. 1968, 49, 219–222. [Google Scholar] [CrossRef]
- Seabright, M. A rapid banding technique for human chromosomes. Lancet 1971, 2, 971–972. [Google Scholar] [CrossRef]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef] [Green Version]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef] [Green Version]
- Trask, B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat. Rev. Genet. 2002, 3, 769–778. [Google Scholar] [CrossRef]
- Bejjani, B.A.; Saleki, R.; Ballif, B.C.; Rorem, E.A.; Sundin, K.; Theisen, A.; Kashork, C.D.; Shaffer, L.G. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: Is less more? Am. J. Med. Genet. 2005, 134, 259–267. [Google Scholar] [CrossRef]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeon, J.T. Methods to detect and analyze copy number variations at the genome-wide and locus-specific levels. Cytogenet. Genome Res. 2008, 123, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Hoebeeck, J.; Speleman, F.; Vandesompele, J. Real-Time Quantitative PCR as an Alternative to Southern Blot or Fluorescence in situ Hybridization for Detection of Gene Copy Number Changes. In Protocols for Nucleic Acid Analysis by Nonradioactive Probes; Humana Press: Totowa, NJ, USA, 2007; pp. 205–226. [Google Scholar]
- Solinas-Toldo, S.; Lampel, S.; Stilgenbauer, S.; Nickolenko, J.; Benner, A.; Döhner, H.; Cremer, T.; Lichter, P. Matrix-based Comparative Genomic Hybridization: Biochips to Screen for Genomic Imbalances. Genes Chromosomes Cancer 1997, 20, 399–407. [Google Scholar] [CrossRef]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.R., 2nd; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, M.S.; Foitzik, M.A.; Surh, L.C.; Korneluk, R.G. Characterization and polymerase chain reaction (PCR) detection of an Alu deletion polymorphism in total linkage disequilibrium with myotonic dystrophy. Genomics 1993, 15, 446–448. [Google Scholar] [CrossRef]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Yau, S.C.; Bobrow, M.; Mathew, C.G.; Abbs, J.S. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J. Med. Genet. 1996, 33, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [Green Version]
- Radvansky, J.; Resko, P.; Surovy, M.; Minarik, G.; Ficek, A.; Kadasi, L. High-resolution melting analysis for genotyping of the myotonic dystrophy type 1 associated Alu insertion/deletion polymorphism. Anal. Biochem. 2010, 398, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazaika, E.; Homsy, J. Digital Droplet PCR: CNV Analysis and Other Applications. Curr. Protoc. Hum. Genet. 2014, 82, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Armour, J.A.; Sismani, C.; Patsalis, P.C.; Cross, G. Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res. 2000, 28, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Patsalis, P.C.; Kousoulidou, L.; Sismani, C.; Männik, K.; Kurg, A. MAPH: From gels to microarrays. Eur. J. Med. Genet. 2005, 48, 241–249. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- DNA Sequencing Costs: Data. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 21 September 2020).
- Wang, H.; Nettleton, D.; Ying, K. Copy number variation detection using next generation sequencing read counts. BMC Bioinform. 2014, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef] [Green Version]
- Magi, A.; Bolognini, D.; Bartalucci, N.; Mingrino, A.; Semeraro, R.; Giovannini, L.; Bonifacio, S.; Parrini, D.; Pelo, E.; Mannelli, F.; et al. Nano-GLADIATOR: Real-time detection of copy number alterations from nanopore sequencing data. Bioinformatics 2019, 35, 4213–4221. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas Lledó, J.I.; Cáceres, M. On the power and the systematic biases of the detection of chromosomal inversions by paired-end genome sequencing. PLoS ONE 2013, 8, e61292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buermans, H.P.J.; Den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Zhang, C.; Li, D.; Carvalho, C.M.B.; Ji, H.; Xiao, J.; Wu, Y.; Zhou, W.; Wang, H.; et al. Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum. Mol. Genet. 2017, 26, 1927–1941. [Google Scholar] [CrossRef]
- Kubiritova, Z.; Gyuraszova, M.; Nagyova, E.; Hyblova, M.; Harsanyova, M.; Budis, J.; Hekel, R.; Gazdarica, J.; Duris, F.; Kadasi, L.; et al. On the critical evaluation and confirmation of germline sequence variants identified using massively parallel sequencing. J. Biotechnol. 2019, 298, 64–75. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Kucharik, M.; Gnip, A.; Hyblova, M.; Budis, J.; Strieskova, L.; Harsanyova, M.; Pös, O.; Kubiritova, Z.; Radvanszky, J.; Minarik, G.; et al. Non-invasive prenatal testing (NIPT) by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions. PLoS ONE 2020, 15, e0238245. [Google Scholar] [CrossRef]
- Straver, R.; Sistermans, E.A.; Holstege, H.; Visser, A.; Oudejans, C.B.M.; Reinders, M.J.T. WISECONDOR: Detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2013, 42, e31. [Google Scholar] [CrossRef]
- Raman, L.; Dheedene, A.; De Smet, M.; Van Dorpe, J.; Menten, B. WisecondorX: Improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2018, 47, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Sathirapongsasuti, J.F.; Lee, H.; Horst, B.A.J.; Brunner, G.; Cochran, A.J.; Binder, S.; Quackenbush, J.; Nelson, S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 2011, 27, 2648–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laver, T.W.; De Franco, E.; Johnson, M.B.; Patel, K.; Ellard, S.; Weedon, M.N.; Flanagan, S.E.; Wakeling, M.N. SavvyCNV: Genome-wide CNV calling from off-target reads. BioRxiv 2019, 617605. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Velds, A.; Kemper, K.; Ranzani, M.; Bombardelli, L.; Hoogstraat, M.; Nevedomskaya, E.; Xu, G.; De Ruiter, J.; Lolkema, M.P.; et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Hunter Shain, A.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Ivakhno, S.; Roller, E.; Colombo, C.; Tedder, P.; Cox, A.J. Canvas SPW: Calling de novo copy number variants in pedigrees. Bioinformatics 2018, 34, 516–518. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, T.; Li, J.; Liu, G.; Yuan, X. MFCNV: A New Method to Detect Copy Number Variations from Next-Generation Sequencing Data. Front. Genet. 2020, 11, 434. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, K.C.; Li, J.; Hunter, S.M.; Ryland, G.L.; Cowin, P.A.; Campbell, I.G.; Halgamuge, S.K. Inferring copy number and genotype in tumour exome data. BMC Genom. 2014, 15, 732. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Hampton, O.; Coarfa, C.; Milosavljevic, A. ReadDepth: A parallel R package for detecting copy number alterations from short sequencing reads. PLoS ONE 2011, 6, e16327. [Google Scholar] [CrossRef] [Green Version]
- Chrisamiller Chrisamiller/Copycat. Available online: https://github.com/chrisamiller/copyCat (accessed on 6 January 2021).
- Yuan, X.; Bai, J.; Zhang, J.; Yang, L.; Duan, J.; Li, Y.; Gao, M. CONDEL: Detecting Copy Number Variation and Genotyping Deletion Zygosity from Single Tumor Samples Using Sequence Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yu, J.; Xi, J.; Yang, L.; Shang, J.; Li, Z.; Duan, J. CNV_IFTV: An isolation forest and total variation-based detection of CNVs from short-read sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Tattini, L.; Cifola, I.; D’Aurizio, R.; Benelli, M.; Mangano, E.; Battaglia, C.; Bonora, E.; Kurg, A.; Seri, M.; et al. EXCAVATOR: Detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013, 14, R120. [Google Scholar] [CrossRef] [Green Version]
- D’Aurizio, R.; Pippucci, T.; Tattini, L.; Giusti, B.; Pellegrini, M.; Magi, A. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 2016, 44, e154. [Google Scholar] [CrossRef]
- Magi, A.; Pippucci, T.; Sidore, C. XCAVATOR: Accurate detection and genotyping of copy number variants from second and third generation whole-genome sequencing experiments. BMC Genom. 2017, 18, 747. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Tammimies, K.; Karlsson, R.; Lu, Y.; Larsson, H.; Lichtenstein, P.; Magnusson, P.K.E. Copy number variation and neuropsychiatric problems in females and males in the general population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.L.; Wain, K.E.; Oetjens, M.T.; Tolwinski, K.; Palen, E.; Hare-Harris, A.; Habegger, L.; Maxwell, E.K.; Reid, J.G.; Walsh, L.K.; et al. Identification of Neuropsychiatric Copy Number Variants in a Health Care System Population. JAMA Psychiatry 2020. [Google Scholar] [CrossRef]
- Park, K.-B.; Nam, K.E.; Cho, A.-R.; Jang, W.; Kim, M.; Park, J.H. Effects of Copy Number Variations on Developmental Aspects of Children with Delayed Development. Ann. Rehabil. Med. 2019, 43, 215–223. [Google Scholar] [CrossRef]
- Glessner, J.T.; Li, J.; Desai, A.; Palmer, M.; Kim, D.; Lucas, A.M.; Chang, X.; Connolly, J.J.; Almoguera, B.; Harley, J.B.; et al. CNV Association of Diverse Clinical Phenotypes from eMERGE reveals novel disease biology underlying cardiovascular disease. Int. J. Cardiol. 2020, 298, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestes, P.R.; Maier, M.C.; Charchar, F.J. DNA copy number variations—Do these big mutations have a big effect on cardiovascular risk? Int. J. Cardiol. 2020, 298, 116–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhang, J.; Liao, D.; Yang, L.; Wang, Y.; Hou, S. Association between C4, C4A, and C4B copy number variations and susceptibility to autoimmune diseases: A meta-analysis. Sci. Rep. 2017, 7, 42628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, K.M.C.; Perazzio, S.; Faria, A.G.A.; Moreira, E.S.; Santos, V.C.; Grecco, M.; Da Silva, N.P.; Andrade, L.E.C. Impact of C4, C4A and C4B gene copy number variation in the susceptibility, phenotype and progression of systemic lupus erythematosus. Adv. Rheumatol. 2019, 59, 36. [Google Scholar] [CrossRef]
- Szilágyi, M.; Pös, O.; Márton, É.; Buglyó, G.; Soltész, B.; Keserű, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef]
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Non-invasive detection of genome-wide somatic copy number alterations by liquid biopsies. Mol. Oncol. 2016, 10, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef] [Green Version]
- Pailler, E.; Auger, N.; Lindsay, C.R.; Vielh, P.; Islas-Morris-Hernandez, A.; Borget, I.; Ngo-Camus, M.; Planchard, D.; Soria, J.-C.; Besse, B.; et al. High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1408–1415. [Google Scholar] [CrossRef]
- Pös, Z.; Pös, O.; Styk, J.; Mocova, A.; Strieskova, L.; Budis, J.; Kadasi, L.; Radvanszky, J.; Szemes, T. Technical and Methodological Aspects of Cell-Free Nucleic Acids Analyzes. Int. J. Mol. Sci. 2020, 21, 8634. [Google Scholar] [CrossRef]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.-H.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; Van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Peng, H.; Lu, L.; Zhou, Z.; Liu, J.; Zhang, D.; Nan, K.; Zhao, X.; Li, F.; Tian, L.; Dong, H.; et al. CNV Detection from Circulating Tumor DNA in Late Stage Non-Small Cell Lung Cancer Patients. Genes 2019, 10, 926. [Google Scholar] [CrossRef] [Green Version]
- Kubiritova, Z.; Radvanszky, J.; Gardlik, R. Cell-Free Nucleic Acids and their Emerging Role in the Pathogenesis and Clinical Management of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2019, 20, 3662. [Google Scholar] [CrossRef] [Green Version]
- Pös, O.; Budiš, J.; Szemes, T. Recent trends in prenatal genetic screening and testing. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Pös, O.; Budis, J.; Kubiritova, Z.; Kucharik, M.; Duris, F.; Radvanszky, J.; Szemes, T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 4403. [Google Scholar] [CrossRef] [Green Version]
- Giles, M.E.; Murphy, L.; Krstić, N.; Sullivan, C.; Hashmi, S.S.; Stevens, B. Prenatal cfDNA screening results indicative of maternal neoplasm: Survey of current practice and management needs. Prenat. Diagn. 2017, 37, 126–132. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.C.; Goodrich, J.K.; Youngblut, N.D.; Luque, G.G.; Ruaud, A.; Sutter, J.L.; Waters, J.L.; Shi, Q.; El-Hadidi, M.; Johnson, L.M.; et al. Human Salivary Amylase Gene Copy Number Impacts Oral and Gut Microbiomes. Cell Host. Microbe. 2019, 25, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Greenblum, S.; Carr, R.; Borenstein, E. Extensive strain-level copy-number variation across human gut microbiome species. Cell 2015, 160, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.M.; McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gaziova, M.; Pos, O.; Krampl, W.; Kubiritova, Z.; Kucharik, M.; Radvanszky, J.; Budis, J.; Szemes, T. Automated prediction of the clinical impact of structural copy number variations. Bioinformatics 2020, 646. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Dharanipragada, P.; Vogeti, S.; Parekh, N. iCopyDAV: Integrated platform for copy number variations—Detection, annotation and visualization. PLoS ONE 2018, 13, e0195334. [Google Scholar] [CrossRef] [Green Version]
- AluScanCNV2: An R package for copy number variation calling and cancer risk prediction with next-generation sequencing data. Genes Dis. 2019, 6, 43–46. [CrossRef]
- Zhao, M.; Zhao, Z. CNVannotator: A Comprehensive Annotation Server for Copy Number Variation in the Human Genome. PLoS ONE 2013, 8, e80170. [Google Scholar] [CrossRef]
- Samarakoon, P.S.; Sorte, H.S.; Stray-Pedersen, A.; Rødningen, O.K.; Rognes, T.; Lyle, R. cnvScan: A CNV screening and annotation tool to improve the clinical utility of computational CNV prediction from exome sequencing data. BMC Genom. 2016, 17, 51. [Google Scholar] [CrossRef] [Green Version]
- Markham, J.F.; Yerneni, S.; Ryland, G.L.; Leong, H.S.; Fellowes, A.; Thompson, E.R.; De Silva, W.; Kumar, A.; Lupat, R.; Li, J.; et al. CNspector: A web-based tool for visualisation and clinical diagnosis of copy number variation from next generation sequencing. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Collins, R.L.; Stone, M.R.; Brand, H.; Glessner, J.T.; Talkowski, M.E. CNView: A visualization and annotation tool for copy number variation from whole-genome sequencing. bioRxiv 2016, 049536. [Google Scholar] [CrossRef] [Green Version]
- Ganel, L.; Abel, H.J.; Hall, I.M. SVScore: An impact prediction tool for structural variation. Bioinformatics 2017, 33, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Erikson, G.A.; Deshpande, N.; Kesavan, B.G.; Torkamani, A. SG-ADVISER CNV: Copy-number variant annotation and interpretation. Genet. Med. 2015, 17, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Spector, J.D.; Wiita, A.P. ClinTAD: A tool for copy number variant interpretation in the context of topologically associated domains. J. Hum. Genet. 2019, 64, 437–443. [Google Scholar] [CrossRef]
- Dalgleish, J.L.; Wang, Y.; Zhu, J.; Meltzer, P.S. CNVScope: Visually Exploring Copy Number Aberrations in Cancer Genomes. Cancer Inform. 2019, 18, 1176935119890290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, Z.; Ban, R.; Zhang, H.; Iqbal, F.; Zhao, A.; Li, A.; Shi, Q. DeAnnCNV: A tool for online detection and annotation of copy number variations from whole-exome sequencing data. Nucleic Acids Res. 2015, 43, W289–W294. [Google Scholar] [CrossRef]
- Gurbich, T.A.; Ilinsky, V.V. ClassifyCNV: A tool for clinical annotation of copy-number variants. Sci. Rep. 2020, 10, 20375. [Google Scholar] [CrossRef]
- Acmg Board of Directors. Laboratory and clinical genomic data sharing is crucial to improving genetic health care: A position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 721–722. [Google Scholar] [CrossRef]


{kind=link}
| Tool | Description | Operating System | Availability | Reference |
|---|---|---|---|---|
| Wisecondor WisecondorX | A tool for detecting small sub-chromosomal and chromosomal genetic CNV alterations in fetal DNA using low coverage sequencing of maternal cfDNA. It allows less-invasive detection of chromosomal CNV changes at a resolution comparable to conventional cytogenetic analysis. Moreover, no re-sequence healthy samples are needed for normalization. | Mac OS X Linux | Free for non-commercial use | [53,54] |
| ExomeCNV | ExomeCNV is based on an algorithm using statistics of sequence coverage and B-allele frequencies for CNV and loss of heterozygosity estimation by mapping short sequence reads. ExomeCNV was the first tool implemented to detect CNVs from WES data. | MS Windows Mac OS X Linux | Free-software license | [55] |
| SAvvyCNV | A tool that uses off-target or non-target reads data from targeted panel and exome sequencing to call CNVs genome-wide. SavvyCNV may call CNVs with high precision and recall. | MS Windows Mac OS X Linux | Free-software license | [56] |
| CopywriteR | A tool that can generate high-quality DNA copy number profiles using off-target reads from targeted sequencing data. In addition, CopywriteR allows extracting accurate copy number information without a reference. | MS Windows Mac OS X Linux | Free-software license | [57] |
| DECoN | A fast and accurate tool for exon CNV detection from whole exons in targeted panel analysis, capable of detecting small intra-exon variants. It provides quality checks and visualization to make it suitable for clinical use. | MS Windows Mac OS X Linux | Freely available | [58] |
| CNVkit | A software toolkit for detection, analysis, and visualization of CNVs, able to estimate CNVs and alterations genome-wide from high-throughput sequencing data. It implements a pipeline for CNV detection that takes advantage of both on- and off-target reads and applies a series of corrections to improve copy number calling accuracy. | Mac OS X Linux | Free software licence | [59] |
| Canvas SPW | Canvas SPW (Small Pedigree Workflow) is a tool for CNV calling that serves to identify germline and de novo CNVs from pedigree sequencing data. In addition, it infers genome-wide parameters such as cancer ploidy, purity and heterogeneity. | MS Windows Linux | Free-software license | [60] |
| MFCNV | A computational method that (i) considers the intrinsic correlations among adjacent positions in the genome, (ii) calculates read depth, GC-content bias, base quality, and correlation value for each genome bin, and (iii) trains a neural network algorithm to predict CNVs. | NA | Free-software license | [61] |
| VarScan 2 | Analysis tool for the detection of somatic mutations and CNVs in exome data from tumor-normal pairs. The algorithm reads data from both samples simultaneously; a heuristic and statistical algorithm detects sequence variants and classifies them by somatic status (germline, somatic, or LOH); while a comparison of normalized read depth delineates relative copy number changes. | MS Windows Mac OS X Linux UNIX | Free for non-commercial use | [62] |
| ADTEx | ADTEx (Aberration Detection in Tumour Exome) is a method to infer somatic CNVs and genotypes using WES data from paired tumour/normal samples. The algorithm uses hidden Markov models to predict CNV counts, genotypes, polyploidy, aneuploidy, cell contamination, and baseline shifts. | Linux | Free-software license | [63] |
| ReadDepth | An R package for inferring CNVs from short-read sequencing data. The algorithm uses a statistical model that accounts for overdispersed data and does not require reference sample data. It includes a method for increasing the resolution from low-coverage experiments by utilizing breakpoint information from paired end sequencing to do positional refinement. For calling somatic CNVs from matched tumor/normal pairs, the authors of ReadDepth recommend a copyCat package that is loosely based on readDepth. | MS Windows Mac OS X Linux | Free software licence | [64,65] |
| CONDEL | CONDEL (CONsensus DELeteriousness) is a method for detecting CNVs from single tumor samples using high-throughput sequence data. It utilizes a novel statistic in combination with a peel-off scheme to assess the statistical significance of genome bins, and adopts a Bayesian approach to infer copy number gains, losses, and deletion zygosity based on statistical mixture models. | MS Windows Mac OS X Linux | Freely available | [66] |
| CNV_IFTV | A method that uses a novel isolation forest algorithm and variation-based detection of CNVs from short-read sequencing data. It is a reliable tool even for low-level coverage and tumor purity. | MS Windows Mac OS X Linux | Freely available | [67] |
| Control-FREEC | A tool for detection of copy-number changes and allelic imbalances (including LOH) using deep-sequencing data. Control-FREEC automatically computes, normalizes, and segments copy number and beta allele frequency profiles, then calls CNVs and LOH. The control sample is optional for WGS data but mandatory for WES or targeted sequencing data. | MS Windows Linux | Free software licence | [68] |
| EXCAVATOR EXCAVATOR2 | EXCAVATOR (EXome Copy number Alterations/Variations annotATOR) a tool for the detection of CNVs from WES data combines a three-step normalization procedure with a hidden Markov model algorithm and a calling method that classifies genomic regions into five copy number states. EXCAVATOR2 is an enhanced version of EXCAVATOR. It is a read count based tool that exploits all the reads produced by WES experiments to detect CNVs with a genome-wide resolution. | Mac OS X Linux | Freely available | [69,70] |
| XCAVATOR | A software package for the identification of genomic regions involved in CNVs from short and long reads in whole-genome sequencing experiments. | Mac Linux | Free-software license | [71] |
| Tool | Description | Operating System | Availability | Reference |
|---|---|---|---|---|
| AnnotSV | A standalone program designed for annotating and ranking SVs. The tool compiles functionally, regulatory and clinically relevant information and aims at providing annotations useful to (i) interpret the potential pathogenicity of SVs and (ii) filter out potential false positives. | MS Windows Mac OS X Linux | Free-software license | [97] |
| iCopyDAV | Integrated platform for CNV detection, annotation and visualization enabling the user to identify CNVs in whole-genome NGS data. iCopyDAV consists of seven modules for (i) calculating optimal bin size; (ii) data preparation; (iii) data pre-treatment; (iv) segmentation; (v) variant calling; (vi) CNV annotation; (vii) plotting CNVs across the chromosome. | Mac OS X Linux | Freely available | [98] |
| AluScanCNV2 | An R package for CNV calling and machine learning-based cancer risk prediction with NGS data. It uses Geary–Hinkley transformation-based comparison of the read-depth. | MS Windows Mac OS X Linux | Free-software license | [99] |
| CNVAnnotator | A web service that displays genomic overlaps of the input coordinates with built-in databases of CNVs and SNPs from genome-wide association studies and additional features such as ENCODE regulatory elements, cytobands, segmental duplications, genome fragile sites, pseudogenes, promoters, enhancers, CpG islands, and methylation sites. | MS Windows Mac OS X Linux | Free access Results are free to academic research. Not for profit | [100] |
| cnvScan | A CNV screening and annotation tool to improve the clinical utility of computational CNV prediction from exome sequencing data. The screening step evaluates CNV prediction using quality scores and refines it using an in-house CNV database. The annotation step uses multiple external databases from three groups: gene and functional effect datasets, known CNVs from public databases and clinically significant datasets. | Linux | Free-software license | [101] |
| CNspector | A web-based tool for the visualization and clinical diagnosis of CNVs from NGS data. It represents a multi-scale interactive browser that shows CNVs in the context of other relevant genomic features to enable faster clinical reporting. | MS Windows Mac OS X Linux | Free-software license | [102] |
| CNView | A tool for normalized visualization, statistical scoring and annotation of CNVs from population-scale WGS datasets having six sequential steps: (i) matrix filtering, (ii) matrix compression, (iii) intra-sample normalization, (iv) inter-sample normalization, (v) coverage visualization, and (vi) genome annotation. | MS Windows Mac OS X Linux | Free-software license | [103] |
| SVScore | A VCF annotation tool that predicts the impact of SVs based on SNP pathogenicity scores across relevant genomic intervals for each SV. The tool assigns a very simple aggregate pathogenicity score to an SV based on overlapping SNP pathogenicity scores. Multiple options for aggregation are supported: maximum, sum, mean and mean of the top N scores. | Linux | Free-software license | [104] |
| SG-ADVISER-CNV | A suite (consisting of an annotation pipeline and a Webserver) for CNV detection and interpretation by performing in-depth annotations and functional predictions for CNVs. The tool is designed to allow users with no prior bioinformatics expertise to handle large volumes of CNV data. | MS Windows Mac OS X Linux | NA | [105] |
| ClinTAD | A browser-based tool for quick evaluation of the clinical relevance of a CNV in the context of TADs. It allows to input a chromosome number, genomic coordinates, and phenotypic information and relate this data to nearby TAD boundaries and genes. | MS Windows Mac OS X Linux | Freely available | [106] |
| CNVScope | A tool for CNV relationship data analysis and visualization, allowing users to create interaction maps, discover CNV map domains, annotate gene interactions, and create interactive visualizations of these CNV interaction maps. | MS Windows Mac OS X Linux | Free-software license | [107] |
| DeAnnCNV | A tool for online detection and annotation of CNVs from WES data. It can extract the shared CNVs among multiple samples and also provides supporting information for the detected CNVs and associated genes. | MS Windows Mac OS X Linux | Freely available | [108] |
| ClassifyCNV | An easy-to-use tool that implements the 2019 ACMG classification guidelines to assess CNV pathogenicity. It uses genomic coordinates and CNV type as input and reports a clinical classification for each variant, a classification score breakdown, and a list of genes of potential importance for variant interpretation. | Mac OS X Linux UNIX | Free for academic and research use only | [109] |
| Database | Abbreviation | Description | Link |
|---|---|---|---|
| ClinVar | ClinVar | A freely accessible, public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. | http://www.ncbi.nlm.nih.gov/clinvar/ |
| Database of genomic structural Variation | dbVar | NCBI’s database of human genomic structural variations with size >50 bp including insertions, deletions, duplications, inversions, mobile elements, translocations, and complex variants. | https://www.ncbi.nlm.nih.gov/dbvar/ |
| DatabasE of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources | DECIPHER | An interactive web-based database, which incorporates a suite of tools designed to aid the interpretation of genomic variants. | https://decipher.sanger.ac.uk |
| Database of Genomic Variants | DGV | The database only contains structural variants identified in healthy control samples. | http://dgv.tcag.ca/dgv/app/home |
| The Genome Aggregation Database | gnomAD-SV | An open resource of structural variation for medical and population genetics. The gnomAD structural variant (SV) callset is available via the gnomAD website and integrated directly into the gnomAD Browser. | https://gnomad.broadinstitute.org |
| Catalogue of Somatic Mutations in Cancer | COSMIC | The world’s largest source of expert manually curated somatic mutation information relating to human cancers. The database combines two main types of data: manually curated high precision data and genome-wide screen data, which provide extensive coverage of the cancer genomic landscape from a somatic perspective. | https://cancer.sanger.ac.uk/cosmic |
| The International Genome Sample Resource | IGSR | The International Genome Sample Resource (IGSR) was established to ensure ongoing usability of data generated by the 1000 Genomes Project and to extend the data set. | https://www.internationalgenome.org/home |
| Autism Chromosome Rearrangement Database | ACRD | A collection of hand curated breakpoints and other genomic features related to autism, taken from publicly available literature, databases and unpublished data. The database is continuously updated with information from in-house experimental data as well as data from published research studies. | http://projects.tcag.ca/autism/ |
| The Chromosome Anomaly Collection | NA | This collection contains examples of unbalanced chromosome abnormalities without phenotypic effect. | http://www.ngrl.org.uk/wessex/collection/index.htm |
| Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer | NA | The information in the database relates cytogenetic changes and their genomic consequences, in particular gene fusions, to tumor characteristics, based either on individual cases or associations. | https://mitelmandatabase.isb-cgc.org |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pös, O.; Radvanszky, J.; Styk, J.; Pös, Z.; Buglyó, G.; Kajsik, M.; Budis, J.; Nagy, B.; Szemes, T. Copy Number Variation: Methods and Clinical Applications. Appl. Sci. 2021, 11, 819. https://doi.org/10.3390/app11020819
Pös O, Radvanszky J, Styk J, Pös Z, Buglyó G, Kajsik M, Budis J, Nagy B, Szemes T. Copy Number Variation: Methods and Clinical Applications. Applied Sciences. 2021; 11(2):819. https://doi.org/10.3390/app11020819
Chicago/Turabian StylePös, Ondrej, Jan Radvanszky, Jakub Styk, Zuzana Pös, Gergely Buglyó, Michal Kajsik, Jaroslav Budis, Bálint Nagy, and Tomas Szemes. 2021. "Copy Number Variation: Methods and Clinical Applications" Applied Sciences 11, no. 2: 819. https://doi.org/10.3390/app11020819
APA StylePös, O., Radvanszky, J., Styk, J., Pös, Z., Buglyó, G., Kajsik, M., Budis, J., Nagy, B., & Szemes, T. (2021). Copy Number Variation: Methods and Clinical Applications. Applied Sciences, 11(2), 819. https://doi.org/10.3390/app11020819