
Interaction between Spike Protein of SARS-CoV-2 and Human Virus Receptor ACE2 Using Two-Color Fluorescence Cross-Correlation Spectroscopy



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Preparation of Plasmid DNA
2.2. Cell Culture and Transfection
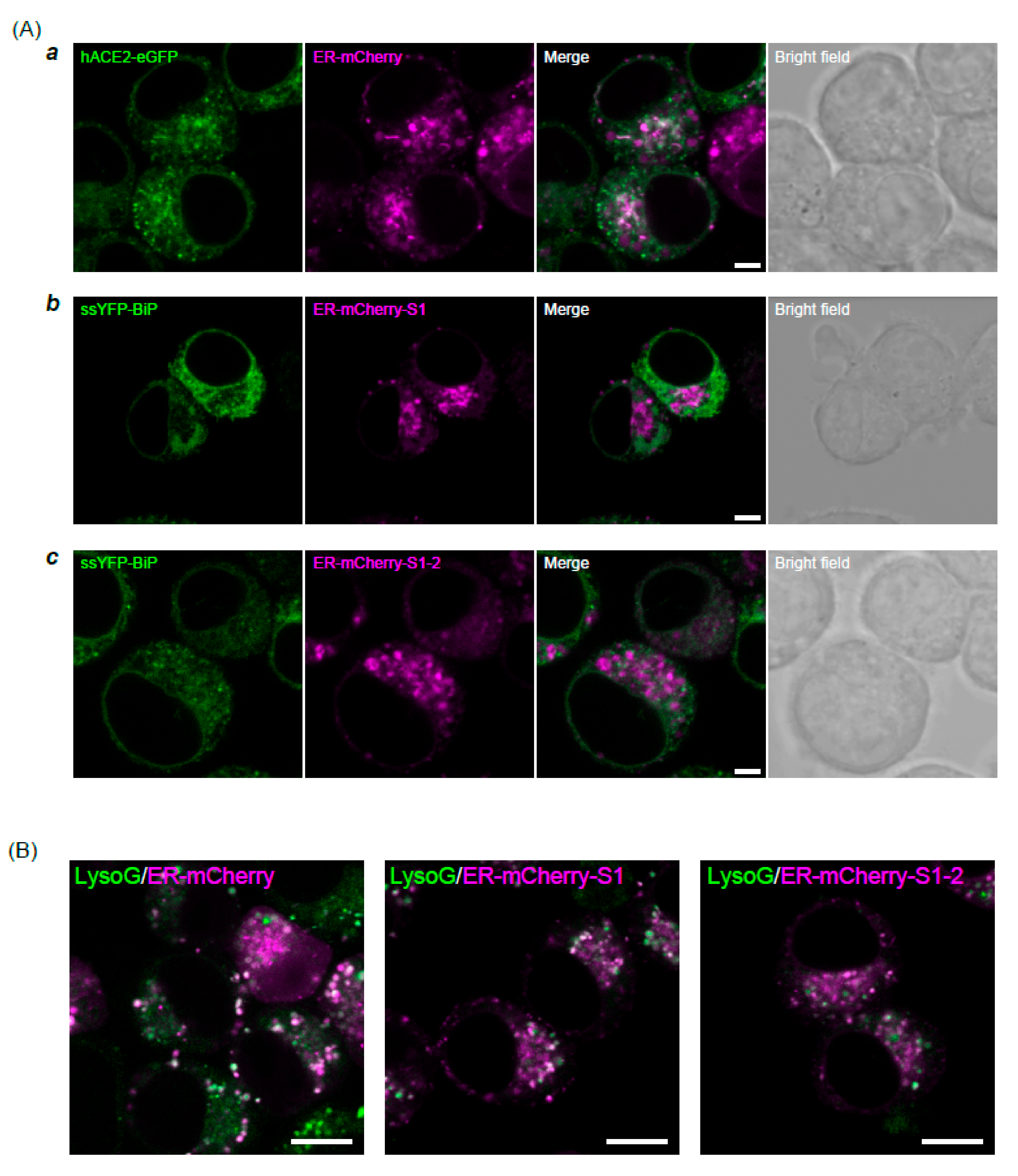
2.3. Confocal Microscopy
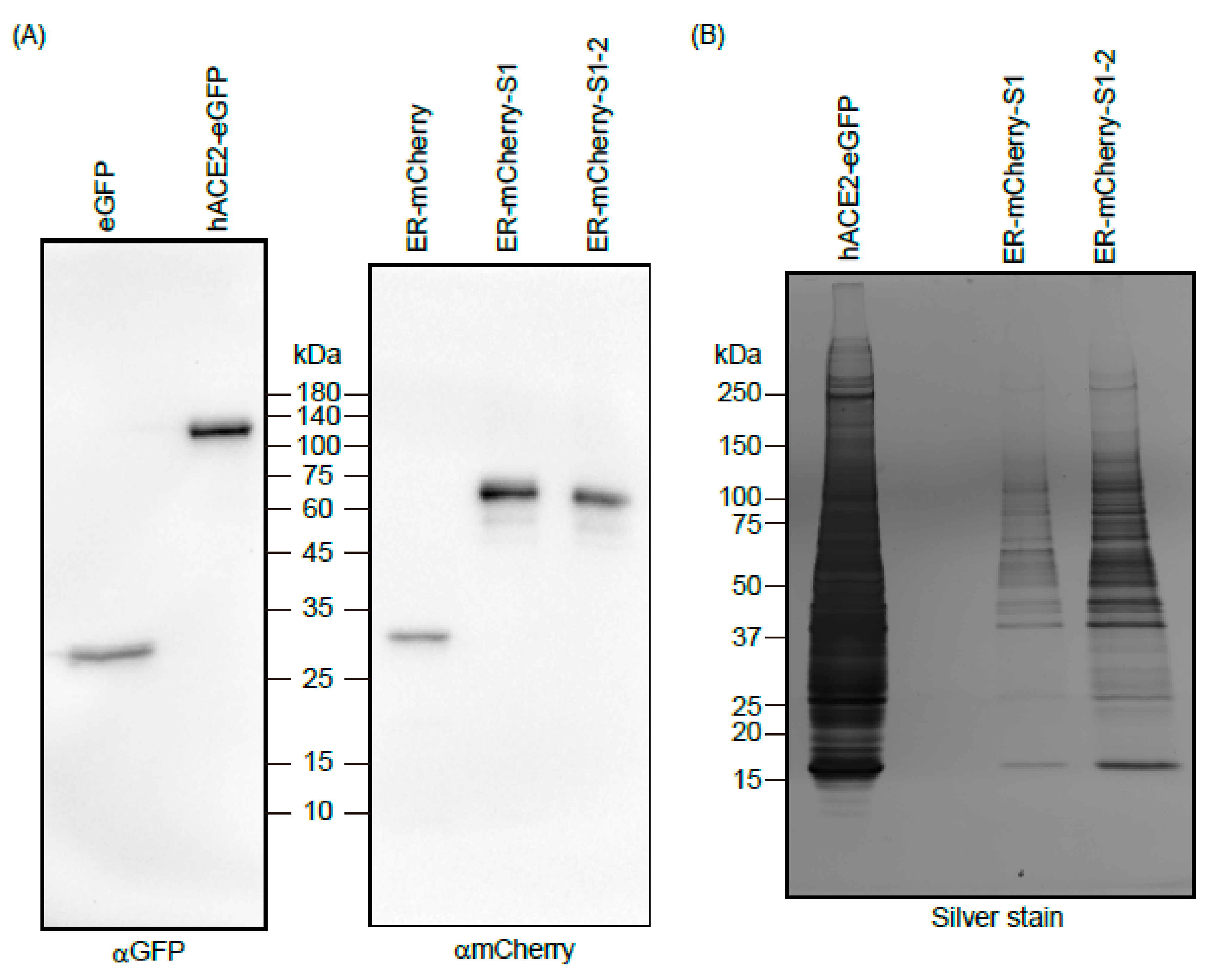
2.4. Protein Purification
2.5. SDS-PAGE and Western Blotting
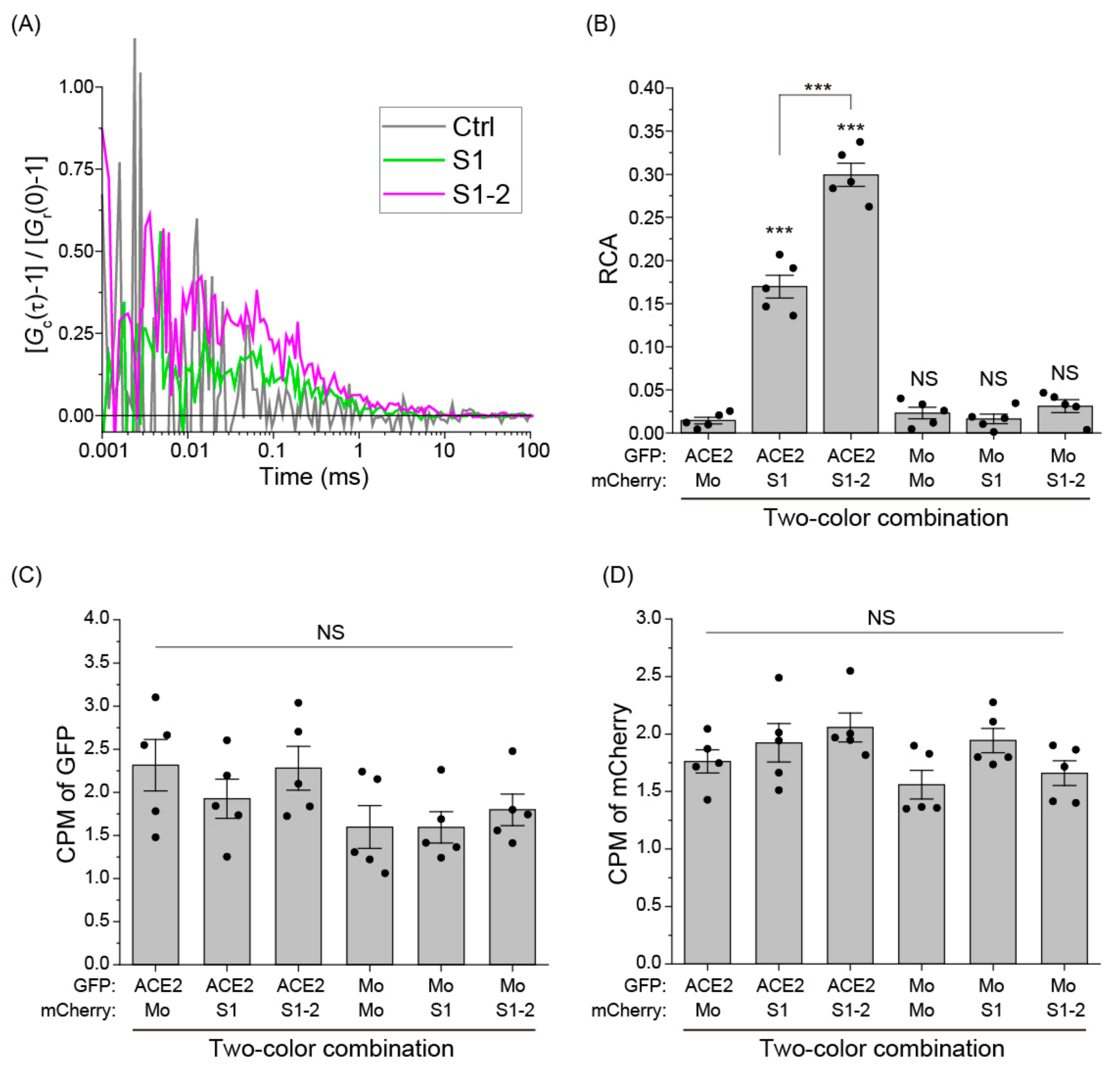
2.6. Fluorescence Cross-Correlation Spectroscopy (FCCS)
2.7. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Zhang, H.; Wada, J.; Hida, K.; Tsuchiyama, Y.; Hiragushi, K.; Shikata, K.; Wang, H.; Lin, S.; Kanwar, Y.S.; Makino, H. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J. Biol. Chem. 2001, 276, 17132–17139. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Su, J.; Lu, H. Opportunities and challenges to the use of neutralizing monoclonal antibody therapies for COVID-19. Biosci. Trends 2021. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.R.; Santos, V.S.; Edwards, T.; Giorgi, E.; Krishna, S.; Planche, T.D.; Staines, H.M.; Fitchett, J.R.A.; Kirwan, D.E.; Cubas Atienzar, A.I.; et al. Prevalence of neutralising antibodies against SARS-CoV-2 in acute infection and convalescence: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2021, 15, e0009551. [Google Scholar] [CrossRef]
- Sadarangani, M.; Marchant, A.; Kollmann, T.R. Immunological mechanisms of vaccine-induced protection against COVID-19 in humans. Nat. Rev. Immunol. 2021, 21, 475–484. [Google Scholar] [CrossRef]
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829. [Google Scholar] [CrossRef]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef]
- Jin, D.; Wei, J.; Sun, J. Analysis of the molecular mechanism of SARS-CoV-2 antibodies. Biochem. Biophys. Res. Commun. 2021, 566, 45–52. [Google Scholar] [CrossRef]
- Hurt, A.C.; Wheatley, A.K. Neutralizing Antibody Therapeutics for COVID-19. Viruses 2021, 13, 628. [Google Scholar] [CrossRef]
- Baral, P.K.; Yin, J.; James, M.N.G. Treatment and prevention strategies for the COVID 19 pandemic: A review of immunotherapeutic approaches for neutralizing SARS-CoV-2. Int. J. Biol. Macromol. 2021, 186, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Vukojevic, V.; Papadopoulos, D.K.; Terenius, L.; Gehring, W.J.; Rigler, R. Quantitative study of synthetic Hox transcription factor-DNA interactions in live cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4093–4098. [Google Scholar] [CrossRef] [Green Version]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Ishida, Y.; Kubota, H.; Pack, C.G.; Homma, T.; Ito, S.; Araki, K.; Kinjo, M.; Nagata, K. Detection of substrate binding of a collagen-specific molecular chaperone HSP47 in solution using fluorescence correlation spectroscopy. Biochem. Biophys. Res. Commun. 2018, 497, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, A.; Kinjo, M. State-of-the-Art Fluorescence Fluctuation-Based Spectroscopic Techniques for the Study of Protein Aggregation. Int. J. Mol. Sci. 2018, 19, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, A.; Yuno, S.; Muto, H.; Kinjo, M. Different aggregation states of a nuclear localization signal-tagged 25-kDa C-terminal fragment of TAR RNA/DNA-binding protein 43 kDa. Genes Cells Devoted Mol. Cell. Mech. 2017, 22, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, A.; Nakayama, Y.; Shibasaki, A.; Taki, A.; Yuno, S.; Takeda, K.; Yahara, M.; Tanabe, N.; Kinjo, M. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci. Rep. 2016, 6, 19230. [Google Scholar] [CrossRef] [Green Version]
- Costantini, L.M.; Baloban, M.; Markwardt, M.L.; Rizzo, M.; Guo, F.; Verkhusha, V.V.; Snapp, E.L. A palette of fluorescent proteins optimized for diverse cellular environments. Nat. Commun. 2015, 6, 7670. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Yamamoto, A.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. GFP-like proteins stably accumulate in lysosomes. Cell Struct. Funct. 2008, 33, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Nagata, K.; Kinjo, M. Conformational analysis of misfolded protein aggregation by FRET and live-cell imaging techniques. Int. J. Mol. Sci. 2015, 16, 6076–6092. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimoto, A.; Lyu, Y.; Kinjo, M.; Kitamura, A. Interaction between Spike Protein of SARS-CoV-2 and Human Virus Receptor ACE2 Using Two-Color Fluorescence Cross-Correlation Spectroscopy. Appl. Sci. 2021, 11, 10697. https://doi.org/10.3390/app112210697
Fujimoto A, Lyu Y, Kinjo M, Kitamura A. Interaction between Spike Protein of SARS-CoV-2 and Human Virus Receptor ACE2 Using Two-Color Fluorescence Cross-Correlation Spectroscopy. Applied Sciences. 2021; 11(22):10697. https://doi.org/10.3390/app112210697
Chicago/Turabian StyleFujimoto, Ai, Yidan Lyu, Masataka Kinjo, and Akira Kitamura. 2021. "Interaction between Spike Protein of SARS-CoV-2 and Human Virus Receptor ACE2 Using Two-Color Fluorescence Cross-Correlation Spectroscopy" Applied Sciences 11, no. 22: 10697. https://doi.org/10.3390/app112210697
APA StyleFujimoto, A., Lyu, Y., Kinjo, M., & Kitamura, A. (2021). Interaction between Spike Protein of SARS-CoV-2 and Human Virus Receptor ACE2 Using Two-Color Fluorescence Cross-Correlation Spectroscopy. Applied Sciences, 11(22), 10697. https://doi.org/10.3390/app112210697