
Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
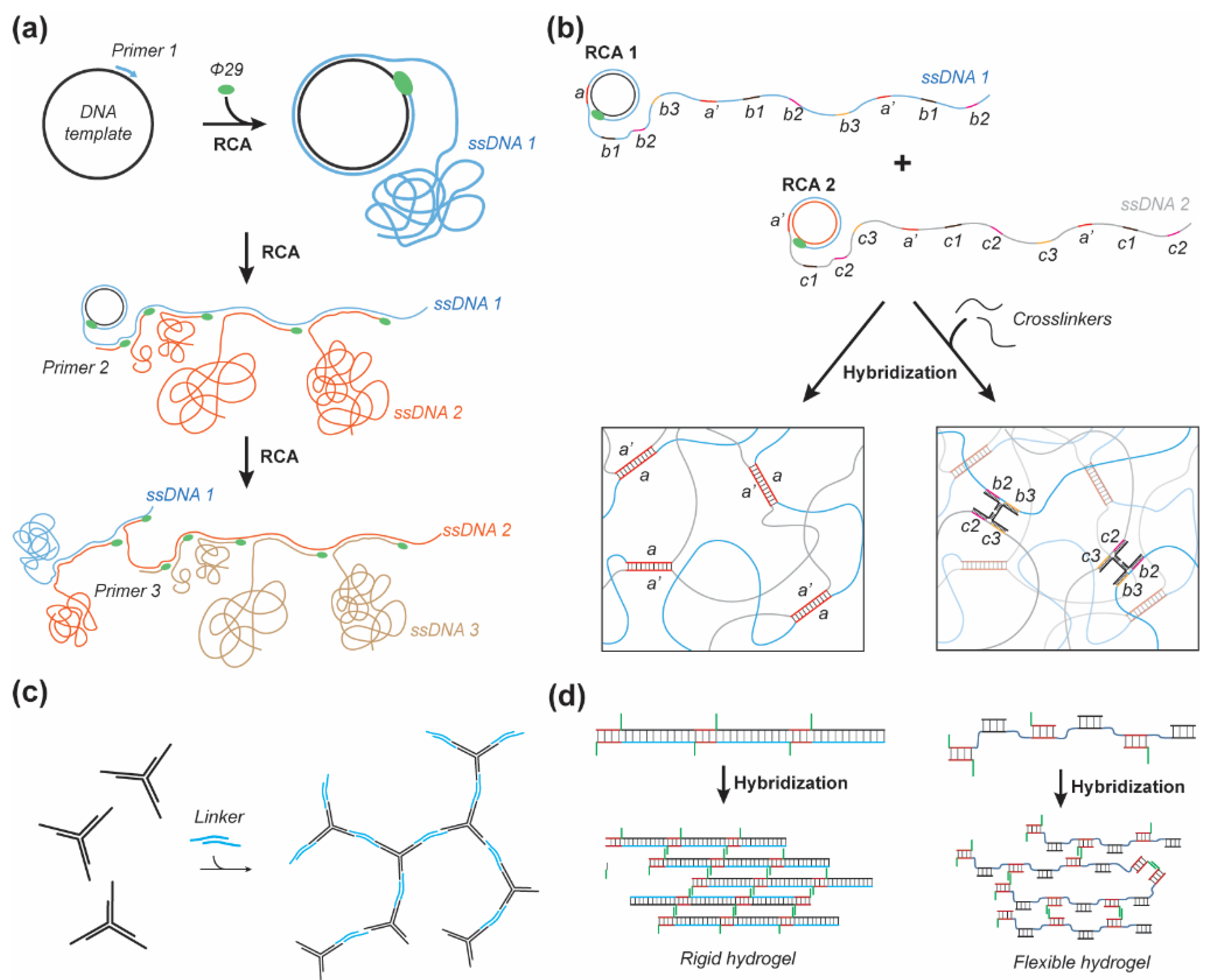
2. Structural DNA Hydrogel Design
2.1. Entangled and Crosslinked
2.2. Nanostructured
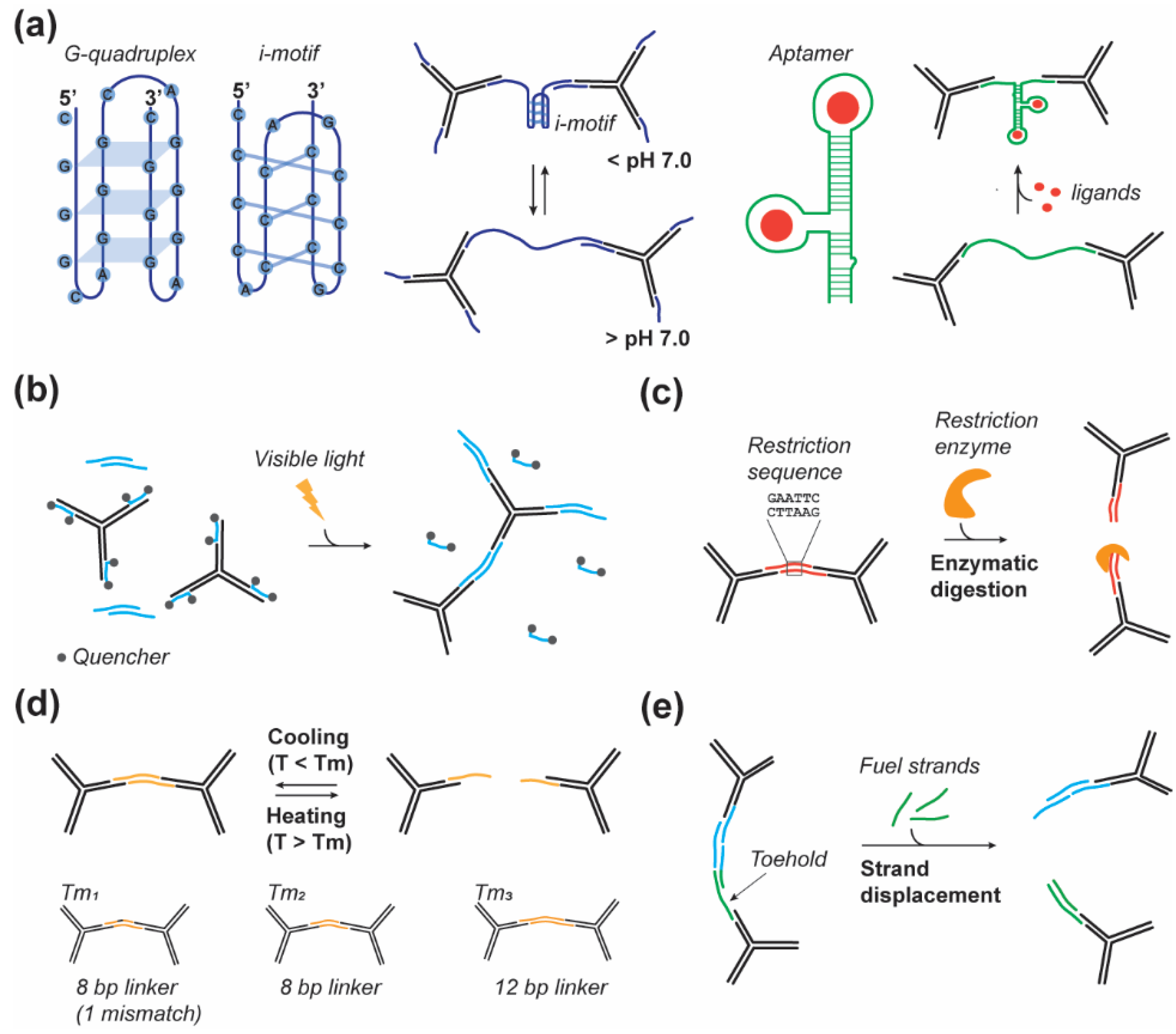
3. Responsive DNA Linkers
3.1. Nucleic Acid Structural Motifs
3.2. Photoresponsive
3.3. Enzyme Responsive
3.4. Thermal Responsive
3.5. Strand Displacement
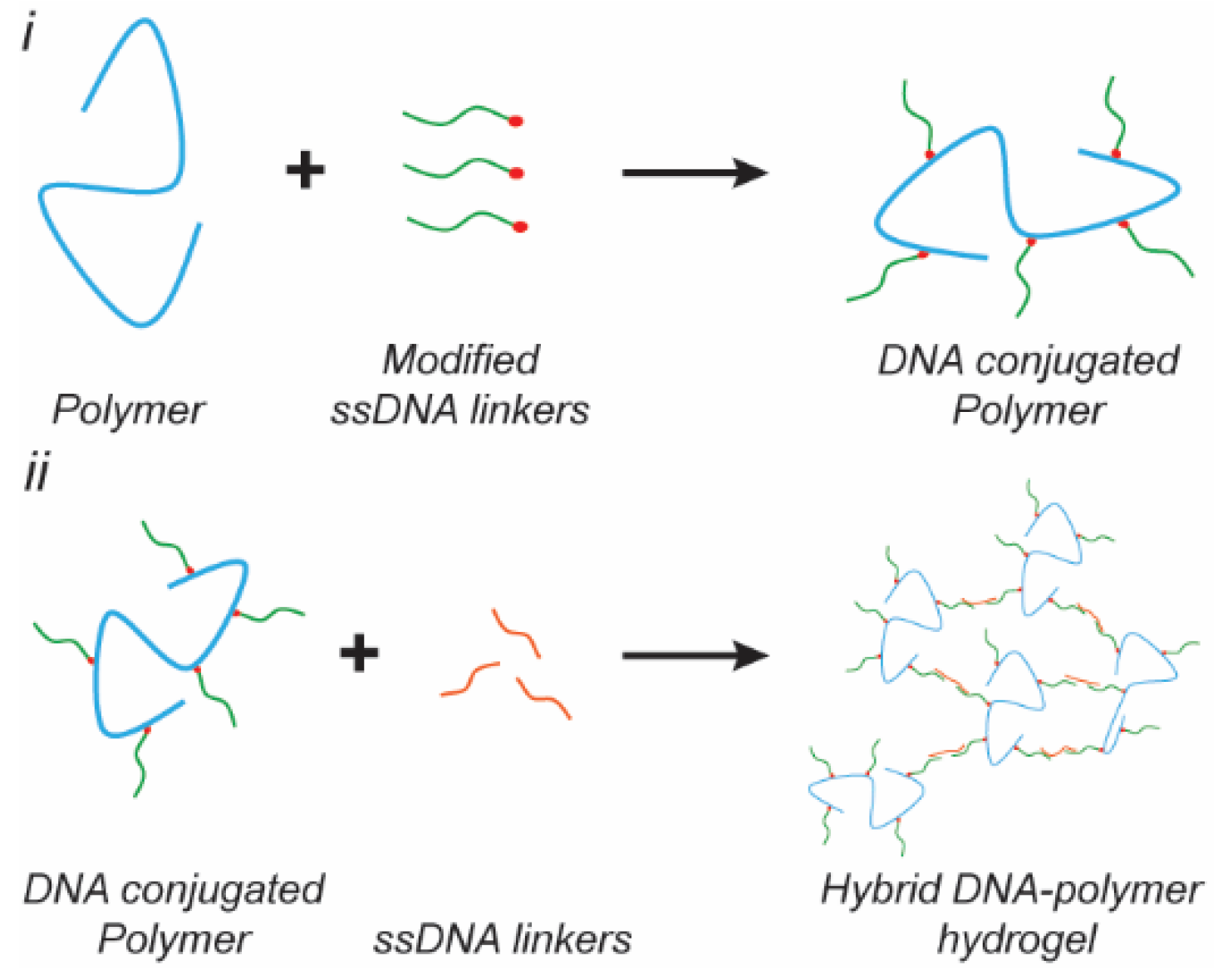
4. DNA–Polymer Hybrids
4.1. Synthetic Polymer Based Hybrids
4.2. Peptide Based Hybrids
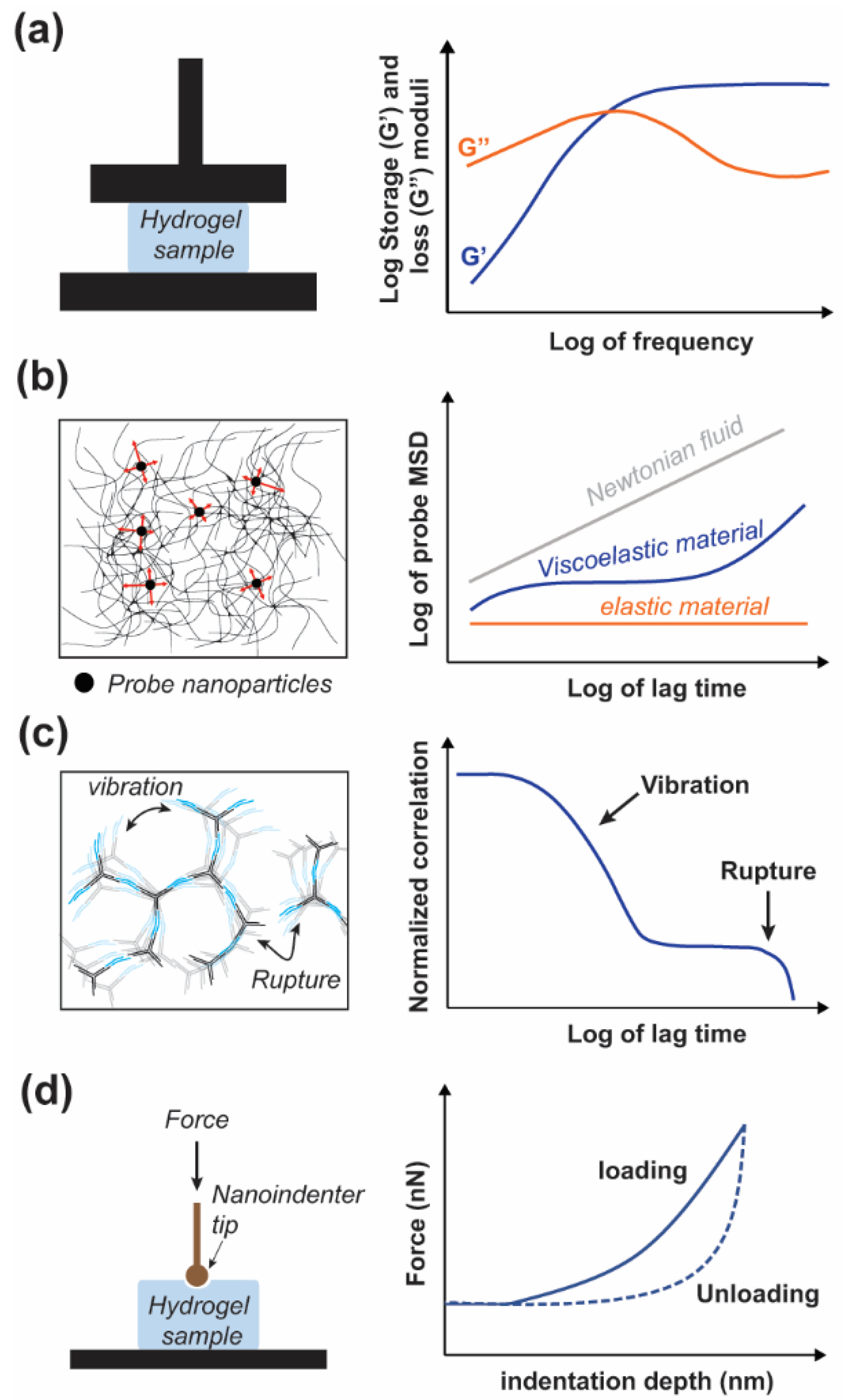
5. Dissecting the Mechanical Properties of DNA Hydrogels
5.1. Characterization Methods
5.2. Modeling/Simulations
5.3. Dynamic Properties of DNA Hydrogels
5.4. Nanoscale Mechanical Properties
6. Mimicking Biomaterials for Biomedical Applications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Seeman, N.C. DNA in a Material World. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. An Overview of Structural DNA Nanotechnology. Mol. Biotechnol. 2007, 37, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and Opportunities for Structural DNA Nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, Y.; Su, M.; Ko, S.H.; Ye, T.; Leng, Y.; Sun, X.; Ribbe, A.E.; Jiang, W.; Mao, C. DNA Self-Assembly: From 2D to 3D. Faraday Discuss. 2009, 143, 221–233. [Google Scholar] [CrossRef]
- Han, D.; Pal, S.; Nangreave, J.; Deng, Z.; Liu, Y.; Yan, H. DNA Origami with Complex Curvatures in Three-Dimensional Space. Science 2011, 332, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneziano, R.; Ratanalert, S.; Zhang, K.; Zhang, F.; Yan, H.; Chiu, W.; Bathe, M. Designer Nanoscale DNA Assemblies Programmed from the Top Down. Science 2016, 352, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Zhang, F.; Liu, Y.; Yan, H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117, 12584–12640. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Y.; Liu, H.; Ribbe, A.E.; Mao, C. Self-Assembly of Hexagonal DNA Two-Dimensional (2D) Arrays. J. Am. Chem. Soc. 2005, 127, 12202–12203. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Mohammed, A.; Gardell, J.; Masich, S.; Czeizler, E.; Orponen, P.; Högberg, B. DNA Rendering of Polyhedral Meshes at the Nanoscale. Nature 2015, 523, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Meyer, T.A.; Pan, V.; Dutta, P.K.; Ke, Y. The Beauty and Utility of DNA Origami. Chem 2017, 2, 359–382. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Castro, C.; Choi, J.H. Structural DNA Nanotechnology: Artificial Nanostructures for Biomedical Research. Annu. Rev. Biomed. Eng. 2018, 20, 375–401. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, M.; Shi, Z.; Castro, C.E.; Arya, G. Dynamic DNA Nanotechnology: Toward Functional Nanoscale Devices. Nanoscale Horiz. 2020, 5, 182–201. [Google Scholar] [CrossRef]
- Chu, T.-W.; Feng, J.; Yang, J.; Kopeček, J. Hybrid Polymeric Hydrogels via Peptide Nucleic Acid (PNA)/DNA Complexation. J. Control. Release 2015, 220, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Lu, C.-H.; Guo, W.; Aleman-Garcia, M.A.; Ren, J.; Willner, I. A Shape Memory Acrylamide/DNA Hydrogel Exhibiting Switchable Dual PH-Responsiveness. Adv. Funct. Mater. 2015, 25, 6867–6874. [Google Scholar] [CrossRef]
- Li, C.; Chen, P.; Shao, Y.; Zhou, X.; Wu, Y.; Yang, Z.; Li, Z.; Weil, T.; Liu, D. A Writable Polypeptide–DNA Hydrogel with Rationally Designed Multi-Modification Sites. Small 2015, 11, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Rowland, M.J.; Shao, Y.; Cao, T.; Chen, C.; Jia, H.; Zhou, X.; Yang, Z.; Scherman, O.A.; Liu, D. Responsive Double Network Hydrogels of Interpenetrating DNA and CB[8] Host–Guest Supramolecular Systems. Adv. Mater. 2015, 27, 3298–3304. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; LaBean, T.H.; Feng, L.; Reif, J.H. Directed Nucleation Assembly of DNA Tile Complexes for Barcode-Patterned Lattices. Proc. Natl. Acad. Sci. USA 2003, 100, 8103–8108. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ye, T.; Su, M.; Zhang, C.; Ribbe, A.E.; Jiang, W.; Mao, C. Hierarchical Self-Assembly of DNA into Symmetric Supramolecular Polyhedra. Nature 2008, 452, 198–201. [Google Scholar] [CrossRef]
- Ong, L.L.; Hanikel, N.; Yaghi, O.K.; Grun, C.; Strauss, M.T.; Bron, P.; Lai-Kee-Him, J.; Schueder, F.; Wang, B.; Wang, P.; et al. Programmable Self-Assembly of Three-Dimensional Nanostructures from 104 Unique Components. Nature 2017, 552, 72–77. [Google Scholar] [CrossRef]
- Jun, H.; Wang, X.; Bricker, W.P.; Bathe, M. Automated Sequence Design of 2D Wireframe DNA Origami with Honeycomb Edges. Nat. Commun. 2019, 10, 5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Marras, A.E.; Su, H.-J.; Castro, C.E. DNA Origami Compliant Nanostructures with Tunable Mechanical Properties. ACS Nano 2014, 8, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.E.; Su, H.-J.; Marras, A.E.; Zhou, L.; Johnson, J. Mechanical Design of DNA Nanostructures. Nanoscale 2015, 7, 5913–5921. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.-J.; Lee, C.; Lee, J.G.; Yagyu, H.; Tabata, O.; Kim, D.-N. Investigating the Sequence-Dependent Mechanical Properties of DNA Nicks for Applications in Twisted DNA Nanostructure Design. Nucleic Acids Res. 2019, 47, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.-H.; Chen, E.; Veneziano, R.; Gaitanaros, S.; Chen, Y. Stretching DNA Origami: Effect of Nicks and Holliday Junctions on the Axial Stiffness. Nucleic Acids Res. 2020, 48, 12407–12414. [Google Scholar] [CrossRef]
- Saran, R.; Wang, Y.; Li, I.T.S. Mechanical Flexibility of DNA: A Quintessential Tool for DNA Nanotechnology. Sensors 2020, 20, 7019. [Google Scholar] [CrossRef]
- Saccà, B.; Meyer, R.; Erkelenz, M.; Kiko, K.; Arndt, A.; Schroeder, H.; Rabe, K.S.; Niemeyer, C.M. Orthogonal Protein Decoration of DNA Origami. Angew. Chem. Int. Ed. Engl. 2010, 49, 9378–9383. [Google Scholar] [CrossRef]
- Wang, P.; Gaitanaros, S.; Lee, S.; Bathe, M.; Shih, W.M.; Ke, Y. Programming Self-Assembly of DNA Origami Honeycomb Two-Dimensional Lattices and Plasmonic Metamaterials. J. Am. Chem. Soc. 2016, 138, 7733–7740. [Google Scholar] [CrossRef]
- Aghebat Rafat, A.; Sagredo, S.; Thalhammer, M.; Simmel, F.C. Barcoded DNA Origami Structures for Multiplexed Optimization and Enrichment of DNA-Based Protein-Binding Cavities. Nat. Chem. 2020, 12, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, X.; Liu, P.; Wang, F.; Ariyama, H.; Ando, T.; Lin, J.; Wang, L.; Hu, J.; Li, B.; et al. Capturing Transient Antibody Conformations with DNA Origami Epitopes. Nat. Commun. 2020, 11, 3114. [Google Scholar] [CrossRef]
- Zhou, X.; Li, C.; Shao, Y.; Chen, C.; Yang, Z.; Liu, D. Reversibly Tuning the Mechanical Properties of a DNA Hydrogel by a DNA Nanomotor. Chem. Commun. 2016, 52, 10668–10671. [Google Scholar] [CrossRef] [PubMed]
- Bohidar, H.; Pandey, P.; Rawat, K.; Vk, A.; Kohlbrecher, J. Structural Hierarchy in DNA Hydrogels. J. Appl. Biotechnol. Bioeng. 2017, 2, 144–150. [Google Scholar]
- Liu, H.; Cao, T.; Xu, Y.; Dong, Y.; Liu, D. Tuning the Mechanical Properties of a DNA Hydrogel in Three Phases Based on ATP Aptamer. Int. J. Mol. Sci. 2018, 19, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gačanin, J.; Synatschke, C.V.; Weil, T. Biomedical Applications of DNA-Based Hydrogels. Adv. Funct. Mater. 2020, 30, 1906253. [Google Scholar] [CrossRef] [Green Version]
- Gačanin, J.; Kovtun, A.; Fischer, S.; Schwager, V.; Quambusch, J.; Kuan, S.L.; Liu, W.; Boldt, F.; Li, C.; Yang, Z.; et al. Spatiotemporally Controlled Release of Rho-Inhibiting C3 Toxin from a Protein–DNA Hybrid Hydrogel for Targeted Inhibition of Osteoclast Formation and Activity. Adv. Healthc. Mater. 2017, 6, 1700392. [Google Scholar] [CrossRef]
- Lyu, D.; Chen, S.; Guo, W. Liposome Crosslinked Polyacrylamide/DNA Hydrogel: A Smart Controlled-Release System for Small Molecular Payloads. Small 2018, 14, 1704039. [Google Scholar] [CrossRef]
- Liwinska, W.; Stanislawska, I.; Lyp, M.; Stojek, Z.; Zabost, E. Switchable Conformational Changes of DNA Nanogel Shells Containing Disulfide–DNA Hybrids for Controlled Drug Release and Efficient Anticancer Action. RSC Adv. 2019, 9, 13736–13748. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Liu, H.; Zhang, X.; Yan, J.; Zhu, Z.; Peng, L.; Yang, H.; Kim, Y.; Tan, W. Photoresponsive DNA-Cross-Linked Hydrogels for Controllable Release and Cancer Therapy. Langmuir 2011, 27, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Prasad Thelu, H.V.; Albert, S.K.; Golla, M.; Krishnan, N.; Ram, D.; Murty Srinivasula, S.; Varghese, R. Size Controllable DNA Nanogels from the Self-Assembly of DNA Nanostructures through Multivalent Host–Guest Interactions. Nanoscale 2018, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Komura, F.; Okuzumi, K.; Takahashi, Y.; Takakura, Y.; Nishikawa, M. Development of RNA/DNA Hydrogel Targeting Toll-Like Receptor 7/8 for Sustained RNA Release and Potent Immune Activation. Molecules 2020, 25, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Ohtsuki, S.; Araie, Y.; Umeki, Y.; Endo, M.; Emura, T.; Hidaka, K.; Sugiyama, H.; Takahashi, Y.; Takakura, Y.; et al. Self-Assembling DNA Hydrogel-Based Delivery of Immunoinhibitory Nucleic Acids to Immune Cells. Nanomedicine 2016, 12, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ishii-Mizuno, Y.; Umeki, Y.; Onuki, Y.; Watanabe, H.; Takahashi, Y.; Takakura, Y.; Nishikawa, M. Improved Sustained Release of Antigen from Immunostimulatory DNA Hydrogel by Electrostatic Interaction with Chitosan. Int. J. Pharm. 2017, 516, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ogawa, K.; Umeki, Y.; Mohri, K.; Kawasaki, Y.; Watanabe, H.; Takahashi, N.; Kusuki, E.; Takahashi, R.; Takahashi, Y.; et al. Injectable, Self-Gelling, Biodegradable, and Immunomodulatory DNA Hydrogel for Antigen Delivery. J. Control. Release 2014, 180, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonášová, E.P.; Stokke, B.T. Bioresponsive DNA-Co-Polymer Hydrogels for Fabrication of Sensors. Curr. Opin. Colloid Interface Sci. 2016, 26, 1–8. [Google Scholar] [CrossRef]
- Li, F.; Lyu, D.; Liu, S.; Guo, W. DNA Hydrogels and Microgels for Biosensing and Biomedical Applications. Adv. Mater. 2019, 32, 1806538. [Google Scholar] [CrossRef]
- Aldaye, F.A.; Senapedis, W.T.; Silver, P.A.; Way, J.C. A Structurally Tunable DNA-Based Extracellular Matrix. J. Am. Chem Soc. 2010, 132, 14727–14729. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Wang, S.; Wulf, V.; Willner, I. Stiffness-Switchable DNA-Based Constitutional Dynamic Network Hydrogels for Self-Healing and Matrix-Guided Controlled Chemical Processes. Nat. Commun. 2019, 10, 4774. [Google Scholar] [CrossRef] [Green Version]
- Merindol, R.; Delechiave, G.; Heinen, L.; Catalani, L.H.; Walther, A. Modular Design of Programmable Mechanofluorescent DNA Hydrogels. Nat. Commun. 2019, 10, 528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.; Xing, Y.; Chen, P.; Yang, Y.; Sun, Y.; Zhou, D.; Xu, L.; Fan, Q.; Liu, D. A PH-Triggered, Fast-Responding DNA Hydrogel. Angew. Chem. Int. Ed. 2009, 48, 7660–7663. [Google Scholar] [CrossRef]
- Um, S.H.; Lee, J.B.; Park, N.; Kwon, S.Y.; Umbach, C.C.; Luo, D. Enzyme-Catalysed Assembly of DNA Hydrogel. Nat. Mater. 2006, 5, 797–801. [Google Scholar] [CrossRef]
- Lee, J.B.; Peng, S.; Yang, D.; Roh, Y.H.; Funabashi, H.; Park, N.; Rice, E.J.; Chen, L.; Long, R.; Wu, M.; et al. A Mechanical Metamaterial Made from a DNA Hydrogel. Nat. Nanotechnol. 2012, 7, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yao, C.; Kou, X.; Tang, J.; Luo, D.; Yang, D. A Fluorescent Biofunctional DNA Hydrogel Prepared by Enzymatic Polymerization. Adv. Healthc. Mater. 2018, 7, 1700998. [Google Scholar] [CrossRef]
- Topuz, F.; Okay, O. Rheological Behavior of Responsive DNA Hydrogels. Macromolecules 2008, 41, 8847–8854. [Google Scholar] [CrossRef]
- Karacan, P.; Cakmak, H.; Okay, O. Swelling Behavior of Physical and Chemical DNA Hydrogels. J. Appl. Polym. Sci. 2013, 128, 3330–3337. [Google Scholar] [CrossRef]
- Basu, S.; Pacelli, S.; Paul, A. Self-Healing DNA-Based Injectable Hydrogels with Reversible Covalent Linkages for Controlled Drug Delivery. Acta Biomater. 2020, 105, 159–169. [Google Scholar] [CrossRef]
- Kosuri, S.; Church, G.M. Large-Scale de Novo DNA Synthesis: Technologies and Applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, F.H.T.; Goossens, E.P.M.; Tessari, M.; Heus, H.A. Enzymatic Preparation of Multimilligram Amounts of Pure Single-Stranded DNA Samples for Material and Analytical Sciences. Anal. Biochem. 2015, 475, 68–73. [Google Scholar] [CrossRef] [PubMed]
- van Emmerik, C.L.; Gachulincova, I.; Lobbia, V.R.; Daniëls, M.A.; Heus, H.A.; Soufi, A.; Nelissen, F.H.T.; van Ingen, H. Ramified Rolling Circle Amplification for Synthesis of Nucleosomal DNA Sequences. Anal. Biochem. 2020, 588, 113469. [Google Scholar] [CrossRef]
- Bush, J.; Singh, S.; Vargas, M.; Oktay, E.; Hu, C.-H.; Veneziano, R. Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25, 3386. [Google Scholar] [CrossRef]
- Lee, C.K.; Shin, S.R.; Lee, S.H.; Jeon, J.-H.; So, I.; Kang, T.M.; Kim, S.I.; Mun, J.Y.; Han, S.-S.; Spinks, G.M.; et al. DNA Hydrogel Fiber with Self-Entanglement Prepared by Using an Ionic Liquid. Angew. Chem. Int. Ed. 2008, 47, 2470–2474. [Google Scholar] [CrossRef]
- Bomboi, F.; Romano, F.; Leo, M.; Fernandez-Castanon, J.; Cerbino, R.; Bellini, T.; Bordi, F.; Filetici, P.; Sciortino, F. Re-Entrant DNA Gels. Nat. Commun. 2016, 7, 13191. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Caciagli, A.; Cao, T.; Stoev, I.; Zupkauskas, M.; O’Neill, T.; Wenzel, T.; Lamboll, R.; Liu, D.; Eiser, E. Microrheology of DNA Hydrogels. Proc. Natl. Acad. Sci. USA 2018, 115, 8137–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, N.; Kennedy, T.; Fygenson, D.K.; Saleh, O.A. Increasing Valence Pushes DNA Nanostar Networks to the Isostatic Point. Proc. Natl. Acad. Sci. USA 2019, 116, 7238–7243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Pan, V.; Vivek, S.; Weeks, E.R.; Ke, Y. Programmable DNA Hydrogels Assembled from Multidomain DNA Strands. ChemBioChem 2016, 17, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Wen, H.; Niu, L.; Su, C.; Liu, C.; Zhao, J.; Mao, C.; Liang, D. Effects of Chain Flexibility on the Properties of DNA Hydrogels. Soft Matter 2016, 12, 5537–5541. [Google Scholar] [CrossRef]
- Stoev, I.D.; Cao, T.; Caciagli, A.; Yu, J.; Ness, C.; Liu, R.; Ghosh, R.; O’Neill, T.; Liu, D.; Eiser, E. On the Role of Flexibility in Linker-Mediated DNA Hydrogels. Soft Matter 2020, 16, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Zhang, Y.; Usdin, K.; Lobachev, K.S. When Secondary Comes First–The Importance of Non-Canonical DNA Structures. Biochimie 2013, 95, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.; Kaushik, S.; Roy, K.; Singh, A.; Mahendru, S.; Kumar, M.; Chaudhary, S.; Ahmed, S.; Kukreti, S. A Bouquet of DNA Structures: Emerging Diversity. Biochem. Biophys. Rep. 2016, 5, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making Sense of G-Quadruplex and i-Motif Functions in Oncogene Promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [Green Version]
- Raiber, E.-A.; Kranaster, R.; Lam, E.; Nikan, M.; Balasubramanian, S. A Non-Canonical DNA Structure Is a Binding Motif for the Transcription Factor SP1 in Vitro. Nucleic Acids Res. 2012, 40, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Brazier, J.A.; Sugimoto, N. Topological Impact of Noncanonical DNA Structures on Klenow Fragment of DNA Polymerase. Proc. Natl. Acad. Sci. USA 2017, 114, 9605–9610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Assi, H.; Garavís, M.; González, C.; Damha, M.J. I-Motif DNA: Structural Features and Significance to Cell Biology. Nucleic Acids Res. 2018, 46, 8038–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikova, S.; Curtis, E.A. Structure and Function of Multimeric G-Quadruplexes. Molecules 2019, 24, 3074. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, M.; Xing, Y.; Li, Y.; Joedecke, C.-C.; Jin, J.; Yang, Z.; Liu, D. Study of PH-Induced Folding and Unfolding Kinetics of the DNA i-Motif by Stopped-Flow Circular Dichroism. Langmuir 2012, 28, 17743–17748. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.R.; Rusling, D.A. Triplex-Forming Oligonucleotides: A Third Strand for DNA Nanotechnology. Nucleic Acids Res. 2018, 46, 1021–1037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, Y.; Ito, Y.; Fujimoto, K. Interstrand Photocrosslinking of DNA via P-Carbamoylvinyl Phenol Nucleoside. Bioorg. Med. Chem. Lett. 2005, 15, 1299–1301. [Google Scholar] [CrossRef]
- Liu, Q.; Deiters, A. Optochemical Control of Deoxyoligonucleotide Function via a Nucleobase-Caging Approach. Acc. Chem. Res. 2014, 47, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, H.; Ito, T.; Yoshida, T.; Liang, X.; Komiyama, M. Photoregulation of the Formation and Dissociation of a DNA Duplex by Using the Cis–Trans Isomerization of Azobenzene. Angew. Chem. Int. Ed. 1999, 38, 2393–2395. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Fujimoto, K. Ultrafast Reversible Photo-Cross-Linking Reaction: Toward in Situ DNA Manipulation. Org. Lett. 2008, 10, 3227–3230. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yoshino, H.; Ami, T.; Yoshimura, Y.; Saito, I. A Light-Controlled Reversible DNA Photoligation via Carbazole-Tethered 5-Carboxyvinyluracil. Org. Lett. 2008, 10, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yamada, A.; Yoshimura, Y.; Tsukaguchi, T.; Sakamoto, T. Details of the Ultrafast DNA Photo-Cross-Linking Reaction of 3-Cyanovinylcarbazole Nucleoside: Cis–Trans Isomeric Effect and the Application for SNP-Based Genotyping. J. Am. Chem. Soc. 2013, 135, 16161–16167. [Google Scholar] [CrossRef] [PubMed]
- Kandatsu, D.; Cervantes-Salguero, K.; Kawamata, I.; Hamada, S.; Nomura, S.M.; Fujimoto, K.; Murata, S. Reversible Gel–Sol Transition of a Photo-Responsive DNA Gel. ChemBioChem 2016, 17, 1118–1121. [Google Scholar] [CrossRef]
- Costa, D.; Valente, A.J.M.; Miguel, M.G.; Lindman, B. Light Triggered Release of Solutes from Covalent DNA Gels. Colloids Surf. A Physicochem. Eng. Asp. 2011, 391, 80–87. [Google Scholar] [CrossRef]
- Costa, D.; Valente, A.J.M.; Queiroz, J. Plasmid DNA Nanogels as Photoresponsive Materials for Multifunctional Bio-Applications. J. Biotechnol. 2015, 202, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, S.; Nishimura, T.; Ogura, Y.; Tanida, J. Photothermal Fabrication of Microscale Patterned DNA Hydrogels. R. Soc. Open Sci. 2018, 5, 171779. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Cheng, E.; Yang, Y.; Chen, P.; Zhang, T.; Sun, Y.; Yang, Z.; Liu, D. Self-Assembled DNA Hydrogels with Designable Thermal and Enzymatic Responsiveness. Adv. Mater. 2011, 23, 1117–1121. [Google Scholar] [CrossRef]
- Ko, O.; Han, S.; Lee, J.B. Selective Release of DNA Nanostructures from DNA Hydrogel. J. Ind. Eng. Chem. 2020, 84, 46–51. [Google Scholar] [CrossRef]
- English, M.A.; Soenksen, L.R.; Gayet, R.V.; de Puig, H.; Angenent-Mari, N.M.; Mao, A.S.; Nguyen, P.Q.; Collins, J.J. Programmable CRISPR-Responsive Smart Materials. Science 2019, 365, 780–785. [Google Scholar] [CrossRef]
- Nagahara, S.; Matsuda, T. Hydrogel Formation via Hybridization of Oligonucleotides Derivatized in Water-Soluble Vinyl Polymers. Polym. Gels Netw. 1996, 4, 111–127. [Google Scholar] [CrossRef]
- Li, C.; Zhou, X.; Shao, Y.; Chen, P.; Xing, Y.; Yang, Z.; Li, Z.; Liu, D. A Supramolecular Hydrogel with Identical Cross-Linking Point Density but Distinctive Rheological Properties. Mater. Chem. Front. 2017, 1, 654–659. [Google Scholar] [CrossRef]
- Fern, J.; Scalise, D.; Cangialosi, A.; Howie, D.; Potters, L.; Schulman, R. DNA Strand-Displacement Timer Circuits. ACS Synth. Biol. 2017, 6, 190–193. [Google Scholar] [CrossRef]
- Cangialosi, A.; Yoon, C.; Liu, J.; Huang, Q.; Guo, J.; Nguyen, T.D.; Gracias, D.H.; Schulman, R. DNA Sequence–Directed Shape Change of Photopatterned Hydrogels via High-Degree Swelling. Science 2017, 357, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Fern, J.; Schulman, R. Modular DNA Strand-Displacement Controllers for Directing Material Expansion. Nat. Commun. 2018, 9, 3766. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, C.; Boldt, F.; Wang, Y.; Kuan, S.L.; Tran, T.T.; Mikhalevich, V.; Förtsch, C.; Barth, H.; Yang, Z.; et al. Programmable Protein–DNA Hybrid Hydrogels for the Immobilization and Release of Functional Proteins. Chem. Commun. 2014, 50, 14620–14622. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Yurke, B.; Langrana, N.A. Mechanical Properties of a Reversible, DNA-Crosslinked Polyacrylamide Hydrogel. J. Biomech. Eng. 2004, 126, 104–110. [Google Scholar] [CrossRef]
- Lin, D.C.; Yurke, B.; Langrana, N.A.; Mills, A.P. A Polyacrylamide Gel With Reversible DNA Crosslinks. Am. Soc. Mech. Eng. Digit. Collect. 2008, 36509, 105–106. [Google Scholar]
- Gupta, A.; Mishra, A.; Puri, N. Peptide Nucleic Acids: Advanced Tools for Biomedical Applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.R.; Hammink, R.; Das, R.K.; Nelissen, F.H.T.; Blank, K.G.; Rowan, A.E.; Heus, H.A. DNA-Responsive Polyisocyanopeptide Hydrogels with Stress-Stiffening Capacity. Adv. Funct. Mater. 2016, 26, 9075–9082. [Google Scholar] [CrossRef]
- Cicuta, P.; Donald, A.M. Microrheology: A Review of the Method and Applications. Soft Matter 2007, 3, 1449–1455. [Google Scholar] [CrossRef]
- Schultz, K.M.; Furst, E.M. Microrheology of Biomaterial Hydrogelators. Soft Matter 2012, 8, 6198–6205. [Google Scholar] [CrossRef]
- Mansel, B.W.; Keen, S.; Patty, P.J.; Hemar, Y.; Williams, M.A.K. A Practical Review of Microrheological Techniques. Rheol. New Concepts Appl. Methods 2013. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wu, C. Rheological Study of Soft Matters: A Review of Microrheology and Microrheometers. Macromol. Chem. Phys. 2018, 219, 1700307. [Google Scholar] [CrossRef]
- Cai, P.C.; Krajina, B.A.; Kratochvil, M.J.; Zou, L.; Zhu, A.; Burgener, E.B.; Bollyky, P.L.; Milla, C.E.; Webber, M.J.; Spakowitz, A.J.; et al. Dynamic Light Scattering Microrheology for Soft and Living Materials. Soft Matter 2020. [Google Scholar] [CrossRef]
- Fernandez-Castanon, J.; Bianchi, S.; Saglimbeni, F.; Leonardo, R.D.; Sciortino, F. Microrheology of DNA Hydrogel Gelling and Melting on Cooling. Soft Matter 2018, 14, 6431–6438. [Google Scholar] [CrossRef] [Green Version]
- Nava, G.; Rossi, M.; Biffi, S.; Sciortino, F.; Bellini, T. Fluctuating Elasticity Mode in Transient Molecular Networks. Phys. Rev. Lett. 2017, 119, 078002. [Google Scholar] [CrossRef]
- Qian, L.; Zhao, H. Nanoindentation of Soft Biological Materials. Micromachines 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, R. Nanomechanical Mapping of Soft Materials with the Atomic Force Microscope: Methods, Theory and Applications. Chem. Soc. Rev. 2020, 49, 5850–5884. [Google Scholar] [CrossRef]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.K.; Louis, A.A. Sequence-Dependent Thermodynamics of a Coarse-Grained DNA Model. J. Chem Phys. 2012, 137, 135101. [Google Scholar] [CrossRef] [PubMed]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing Improved Structural Properties and Salt Dependence into a Coarse-Grained Model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SantaLucia, J.; Allawi, H.T.; Seneviratne, P.A. Improved Nearest-Neighbor Parameters for Predicting DNA Duplex Stability. Biochemistry 1996, 35, 3555–3562. [Google Scholar] [CrossRef]
- Rovigatti, L.; Bomboi, F.; Sciortino, F. Accurate Phase Diagram of Tetravalent DNA Nanostars. J. Chem. Phys. 2014, 140, 154903. [Google Scholar] [CrossRef] [Green Version]
- Rovigatti, L.; Smallenburg, F.; Romano, F.; Sciortino, F. Gels of DNA Nanostars Never Crystallize. ACS Nano 2014, 8, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Smallenburg, F.; Sciortino, F. Liquids More Stable than Crystals in Particles with Limited Valence and Flexible Bonds. Nat. Phys. 2013, 9, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Castanon, J.; Bomboi, F.; Rovigatti, L.; Zanatta, M.; Paciaroni, A.; Comez, L.; Porcar, L.; Jafta, C.J.; Fadda, G.C.; Bellini, T.; et al. Small-Angle Neutron Scattering and Molecular Dynamics Structural Study of Gelling DNA Nanostars. J. Chem. Phys. 2016, 145, 084910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Z.; Ness, C.; Frenkel, D.; Eiser, E. Structural and Linear Elastic Properties of DNA Hydrogels by Coarse-Grained Simulation. Macromolecules 2019, 52, 504–512. [Google Scholar] [CrossRef] [Green Version]
- Biffi, S.; Cerbino, R.; Bomboi, F.; Paraboschi, E.M.; Asselta, R.; Sciortino, F.; Bellini, T. Phase Behavior and Critical Activated Dynamics of Limited-Valence DNA Nanostars. Proc. Natl. Acad. Sci. USA 2013, 110, 15633–15637. [Google Scholar] [CrossRef] [Green Version]
- Biffi, S.; Cerbino, R.; Nava, G.; Bomboi, F.; Sciortino, F.; Bellini, T. Equilibrium Gels of Low-Valence DNA Nanostars: A Colloidal Model for Strong Glass Formers. Soft Matter 2015, 11, 3132–3138. [Google Scholar] [CrossRef]
- Fernandez-Castanon, J.; Bomboi, F.; Sciortino, F. Binding Branched and Linear DNA Structures: From Isolated Clusters to Fully Bonded Gels. J. Chem. Phys. 2018, 148, 025103. [Google Scholar] [CrossRef]
- Romano, F.; Sciortino, F. Switching Bonds in a DNA Gel: An All-DNA Vitrimer. Phys. Rev. Lett. 2015, 114, 078104. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, C.; DeSantis, P.; Scipioni, A. Nanoscale Mechanical and Dynamical Properties of DNA Single Molecules. Biophys. Chem. 2005, 113, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.-A.; Bauleth-Ramos, T.; Santos, H.A. DNA Hydrogel Assemblies: Bridging Synthesis Principles to Biomedical Applications. Adv. Ther. 2018, 1, 1800042. [Google Scholar] [CrossRef]
- Bloom, K.S. Beyond the Code: The Mechanical Properties of DNA as They Relate to Mitosis. Chromosoma 2008, 117, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, C.M.; Ramos, D.; Mendieta-Moreno, J.I.; Fierro, J.L.G.; Mendieta, J.; Tamayo, J.; Calleja, M. Effect of Water-DNA Interactions on Elastic Properties of DNA Self-Assembled Monolayers. Sci. Rep. 2017, 7, 536. [Google Scholar] [CrossRef] [Green Version]
- Maragakis, P.; Barnett, R.L.; Kaxiras, E.; Elstner, M.; Frauenheim, T. Electronic Structure of Overstretched DNA. Phys. Rev. B 2002, 66, 241104. [Google Scholar] [CrossRef]
- Kubař, T.; Elstner, M. What Governs the Charge Transfer in DNA? The Role of DNA Conformation and Environment. J. Phys. Chem. B 2008, 112, 8788–8798. [Google Scholar] [CrossRef]
- Kundu, S.; Karmakar, S.N. Conformation Dependent Electronic Transport in a DNA Double-Helix. AIP Adv. 2015, 5, 107122. [Google Scholar] [CrossRef] [Green Version]
- Salazar, S.V.; Mujica, V.; Medina, E. Spin-Orbit Coupling Modulation in DNA by Mechanical Deformations. CHIMIA Int. J. Chem. 2018, 72, 411–417. [Google Scholar] [CrossRef]
- Thomas, J.M.; Yu, H.-Z.; Sen, D. A Mechano-Electronic DNA Switch. J. Am. Chem. Soc. 2012, 134, 13738–13748. [Google Scholar] [CrossRef]
- Artés, J.M.; Li, Y.; Qi, J.; Anantram, M.P.; Hihath, J. Conformational Gating of DNA Conductance. Nat. Commun. 2015, 6, 8870. [Google Scholar] [CrossRef]
- Wu, Z.-S.; Chen, C.-R.; Shen, G.-L.; Yu, R.-Q. Reversible Electronic Nanoswitch Based on DNA G-Quadruplex Conformation: A Platform for Single-Step, Reagentless Potassium Detection. Biomaterials 2008, 29, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, C.; Zhu, C.; Salaita, K. DNA-Based Digital Tension Probes Reveal Integrin Forces during Early Cell Adhesion. Nat. Commun. 2014, 5, 5167. [Google Scholar] [CrossRef]
- Ma, R.; Kellner, A.V.; Ma, V.P.-Y.; Su, H.; Deal, B.R.; Brockman, J.M.; Salaita, K. DNA Probes That Store Mechanical Information Reveal Transient Piconewton Forces Applied by T Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 16949–16954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, N.; Xie, T.; Bagheri, Y.; Liang, C.; Keshri, P.; Sun, Y.; You, M. Quantifying Tensile Forces at Cell–Cell Junctions with a DNA-Based Fluorescent Probe. Chem. Sci. 2020, 11, 8558–8566. [Google Scholar] [CrossRef]
- Baig, M.M.F.A.; Lai, W.-F.; Akhtar, M.F.; Saleem, A.; Ahmed, S.A.; Xia, X.-H. DNA Nanotechnology as a Tool to Develop Molecular Tension Probes for Bio-Sensing and Bio-Imaging Applications: An up-to-Date Review. Nano-Struct. Nano-Objects 2020, 23, 100523. [Google Scholar] [CrossRef]
- Glazier, R.; Shinde, P.; Ogasawara, H.; Salaita, K. Spectroscopic Analysis of a Library of DNA Tension Probes for Mapping Cellular Forces at Fluid Interfaces. ACS Appl. Mater. Interfaces 2021, 13, 2145–2164. [Google Scholar] [CrossRef]
- Hinderer, S.; Layland, S.L.; Schenke-Layland, K. ECM and ECM-like Materials—Biomaterials for Applications in Regenerative Medicine and Cancer Therapy. Adv. Drug Deliv. Rev. 2016, 97, 260–269. [Google Scholar] [CrossRef]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D Extracellular Matrix Mimics: Fundamental Concepts and Role of Materials Chemistry to Influence Stem Cell Fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef]
- Nishikawa, M.; Mizuno, Y.; Mohri, K.; Matsuoka, N.; Rattanakiat, S.; Takahashi, Y.; Funabashi, H.; Luo, D.; Takakura, Y. Biodegradable CpG DNA Hydrogels for Sustained Delivery of Doxorubicin and Immunostimulatory Signals in Tumor-Bearing Mice. Biomaterials 2011, 32, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.; Kojima, T.; Song, Y.; Takayama, S. DNA-Based Biomaterials for Immunoengineering. Adv. Healthc. Mater. 2019, 8, 1801243. [Google Scholar] [CrossRef]
- Lattuada, E.; Leo, M.; Caprara, D.; Salvatori, L.; Stoppacciaro, A.; Sciortino, F.; Filetici, P. DNA-GEL, Novel Nanomaterial for Biomedical Applications and Delivery of Bioactive Molecules. Front. Pharm. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Patel, K.; Perez-Garrido, S.; Marshall, J.F.; Palma, M. DNA Origami Nanoarrays for Multivalent Investigations of Cancer Cell Spreading with Nanoscale Spatial Resolution and Single-Molecule Control. ACS Nano 2019, 13, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, W.; Huang, D.; Reynolds, P.; Hammond, L.; Ward, M.; Gadegaard, N.; Marshall, J.F.; Iskratsch, T.; Palma, M. Probing the Nanoscale Organisation and Multivalency of Cell Surface Receptors: DNA Origami Nanoarrays for Cellular Studies with Single-Molecule Control. Faraday Discuss. 2019, 219, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Chi, Q.; Yang, Z.; Xu, K.; Wang, C.; Liang, H. DNA Nanostructure as an Efficient Drug Delivery Platform for Immunotherapy. Front. Pharm. 2020, 10. [Google Scholar] [CrossRef]
- Finke, A.; Bußkamp, H.; Manea, M.; Marx, A. Designer Extracellular Matrix Based on DNA–Peptide Networks Generated by Polymerase Chain Reaction. Angew. Chem. Int. Ed. 2016, 55, 10136–10140. [Google Scholar] [CrossRef]
- Jiang, F.X.; Yurke, B.; Firestein, B.L.; Langrana, N.A. Neurite Outgrowth on a DNA Crosslinked Hydrogel with Tunable Stiffnesses. Ann. Biomed. Eng. 2008, 36, 1565. [Google Scholar] [CrossRef]
- Basu, S.; Alkiswani, A.-R.; Pacelli, S.; Paul, A. Nucleic Acid-Based Dual Cross-Linked Hydrogels for in Situ Tissue Repair via Directional Stem Cell Migration. ACS Appl. Mater. Interfaces 2019, 11, 34621–34633. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.S.; Hu, Y.; Willner, I. Stimuli-Responsive DNA-Based Hydrogels: From Basic Principles to Applications. Acc. Chem. Res. 2017, 50, 680–690. [Google Scholar] [CrossRef]
- Bila, H.; Kurisinkal, E.E.; Bastings, M.M.C. Engineering a Stable Future for DNA-Origami as a Biomaterial. Biomater. Sci. 2019, 7, 532–541. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bush, J.; Hu, C.-H.; Veneziano, R. Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials. Appl. Sci. 2021, 11, 1885. https://doi.org/10.3390/app11041885
Bush J, Hu C-H, Veneziano R. Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials. Applied Sciences. 2021; 11(4):1885. https://doi.org/10.3390/app11041885
Chicago/Turabian StyleBush, Joshua, Chih-Hsiang Hu, and Remi Veneziano. 2021. "Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials" Applied Sciences 11, no. 4: 1885. https://doi.org/10.3390/app11041885
APA StyleBush, J., Hu, C. -H., & Veneziano, R. (2021). Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials. Applied Sciences, 11(4), 1885. https://doi.org/10.3390/app11041885