1. Introduction
Cerebellar Atrophy (CA) is a neuroradiological definition for an arborized cerebellum with progressive and irreversible volume reduction, where neurons have been lost following a neurodegenerative process [
1,
2,
3]. Numerous diseases, which especially in children may have a genetic cause, share such a brain-imaging pattern.
When addressing cerebellar genetic diseases in childhood, it is relatively easy to formulate a diagnosis in the case of cerebellar malformations, for which an extensive categorization conceived by Barkovich in 2009 serves as an accurate guide [
4]. The diagnostic process is more challenging for cerebellar atrophy, as the radiological patterns are often overlapping, tend to evolve over time, and it is often not possible reach an etiopathological definition based solely on brain Magnetic Resonance Imaging (MRI) findings. The most complete and recent classification of cerebellar degenerative patterns was published by Poretti and colleagues and it is essentially based on neuroimaging [
3].
CA can affect the vermis, the hemispheres, or both, but most of the time the vermis is more severely affected [
2]. A peculiar hypo-atrophic pattern, where atrophy is superimposed on hypoplasia, characterizes diseases caused by genes responsible for cerebellar integrity and trophism; these genes are not primarily implicated in embryonic brain development, but the detrimental insult is early and severe, causing in utero damage mimicking malformative disease. This is mainly typical of Congenital Disorders of Glycosylation (CDG) and Pontocerebellar Hypoplasias (PCH), where the severity of the atrophy may or may not evolve [
2,
4].
Despite the miscellaneous MRI patterns and the variety of the genetic pathways involved, the clinical presentation can be very similar among patients. Initially, developmental stages may be normally achieved, but unbalance and incoordination with gait, stance, and sitting problems emerge in most children as a major sign of cerebellar involvement. Many pediatric patients also show a reduced muscular tone, even in the absence of pyramidal or peripheral nervous system involvement, confirming the role of the cerebellum in tone maintenance. Nystagmus, saccadic intrusion, and impaired pursuit are frequently found, as well as dysarthria and orobuccal dyspraxia. Finally, a global developmental delay is often present, confirming the cerebellum’s role in cognition (reviewed by Bodranghien et al. [
5]).
Beyond cerebellar involvement, children with CA may present signs of the impairment of other neurological or extra-neurological systems. Brain MRI can reveal the concomitant atrophy of supratentorial regions, more typically in ravaging disorders with severe and progressive disability, as in the case of Neuronal Ceroid Lipofuscinosis (NCL); white matter (WM) abnormalities may be found, and that is the case of the Hypomyelinating Leukodystrophies (HLD) with cerebellar involvement, but also in some DNA repair defect disorders [
2,
6]. This latter group of diseases typically show extrapyramidal system deterioration and Peripheral Nervous System (PNS) involvement (axonal neuropathy). The concurrent presence of cerebellar and pyramidal signs is instead more common in some Spinocerebellar Ataxias (SCA) or metabolic/mitochondrial disorders [
6,
7]. Retinopathy can be present, but it is not immediately attributable to a specific disorder, although it may hint at SCA or mitochondrial diseases [
8,
9].
Given the unspecific clinical presentation, overlapping radiological patterns, and shared comorbidities, the diagnosis of children with CA is often challenging. In more recent years, the progress of next-generation sequencing (NGS)-based techniques has significantly increased the diagnostic yield of pediatric ataxias. However, the broader the analysis is, the more variants are detected and need interpretation, and only a definite knowledge of the clinical presentation may help in reaching a molecular diagnosis. Precise phenotype definition is indeed essential, also because ataxias may follow non-Mendelian patterns of inheritance (e.g., nucleotide expansions, mitochondrial) and because null alleles can derive from a partial or complete loss of the gene: such assets can be lost with NGS-based diagnostic strategies and require a precise suspicion to be tested for.
In the medical literature, a number of diagnostic algorithms for cerebellar degenerative disorders in childhood [
2,
3,
6,
8,
9] have been proposed based on neuroradiology or diseases’ description in the medical literature, with a limited participation of clinics in the process of disease definition.
Here, we report our experience with CA: 80 pediatric patients with radiological evidence of CA were recruited; in 52 a precise disease categorization was possible and in 79% of this subgroup a genetic confirmation was also obtained. We describe the tailored process leading to diagnosis in such patients, proposing a newly conceived diagnostic approach to cerebellar atrophy in children, with a focus on clinical diagnostic handles.
3. Results
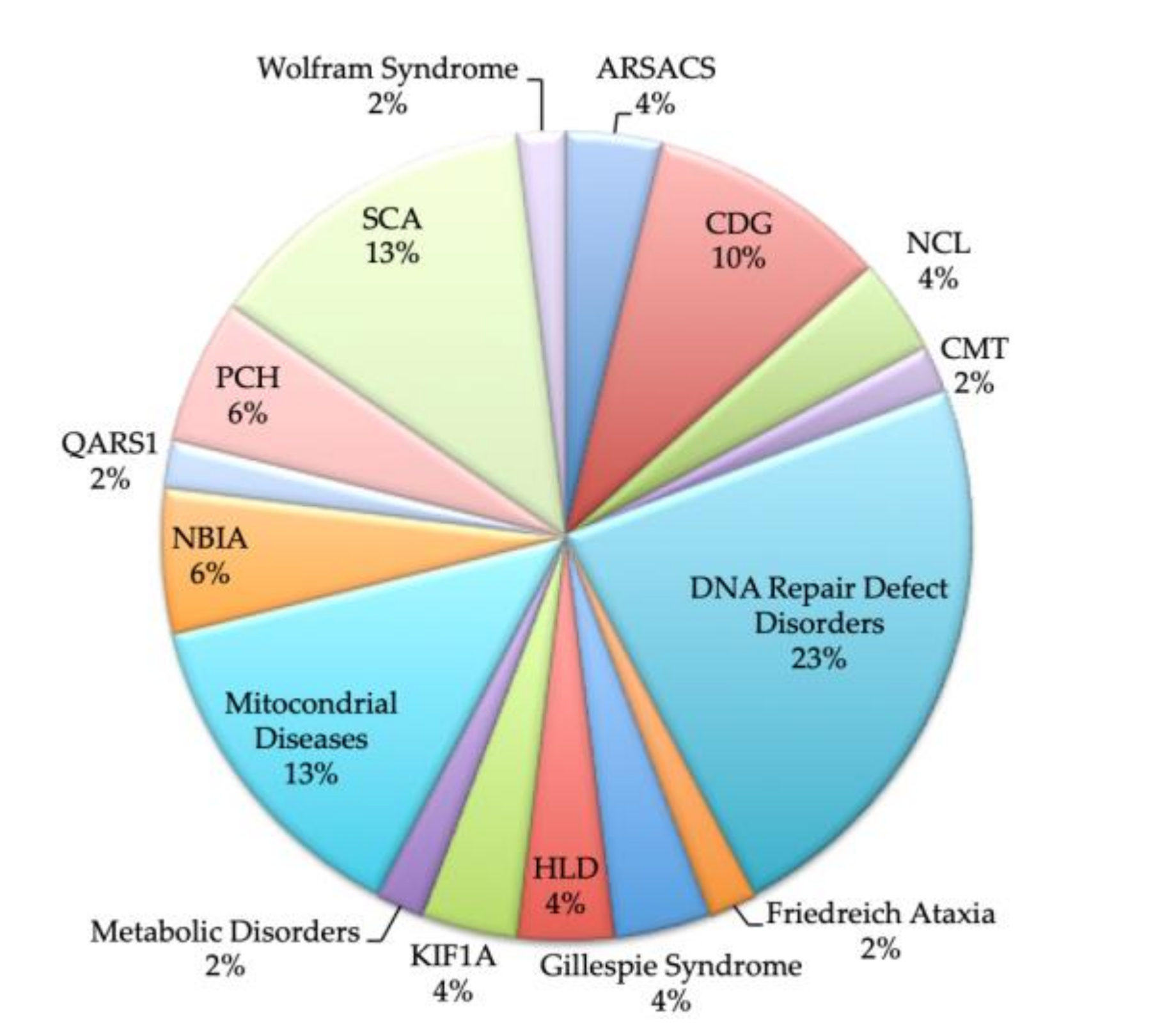
The diagnostic rate in the original 80-patient cohort was 65%. The distribution of diagnosis is shown in
Figure 1. Of the 52 patients, a genetic confirmation was obtained in 79% (41/52). The patients lacking a molecular diagnosis include three CDG patients who have a confirmed Pattern I at the T-IEF (in two of them, brothers, a single mutation in ALG9 was found), two with Cockayne, who only have confirmation on fibroblasts, and six patients with urinary OA alterations and/or mitochondrial studies alterations in which the molecular cause is missing despite extensive genetic analyses.
In the 52-patient cohort, the male to female ratio was 0.68 (M 21/F 31). Age at first diagnosis ranged from 3 months (3 m) to 13 years and 3 months (13 y5 m), with a mean of 5 y4 m and a median of 4 y6 m.
Table A1 summarizes the prevalence of the main clinical findings, while
Table A2 lists the neurophysiological and laboratory exam results; complete data about personal information, clinical features, and exam results are reported in
Supplementary Material, Table S1.
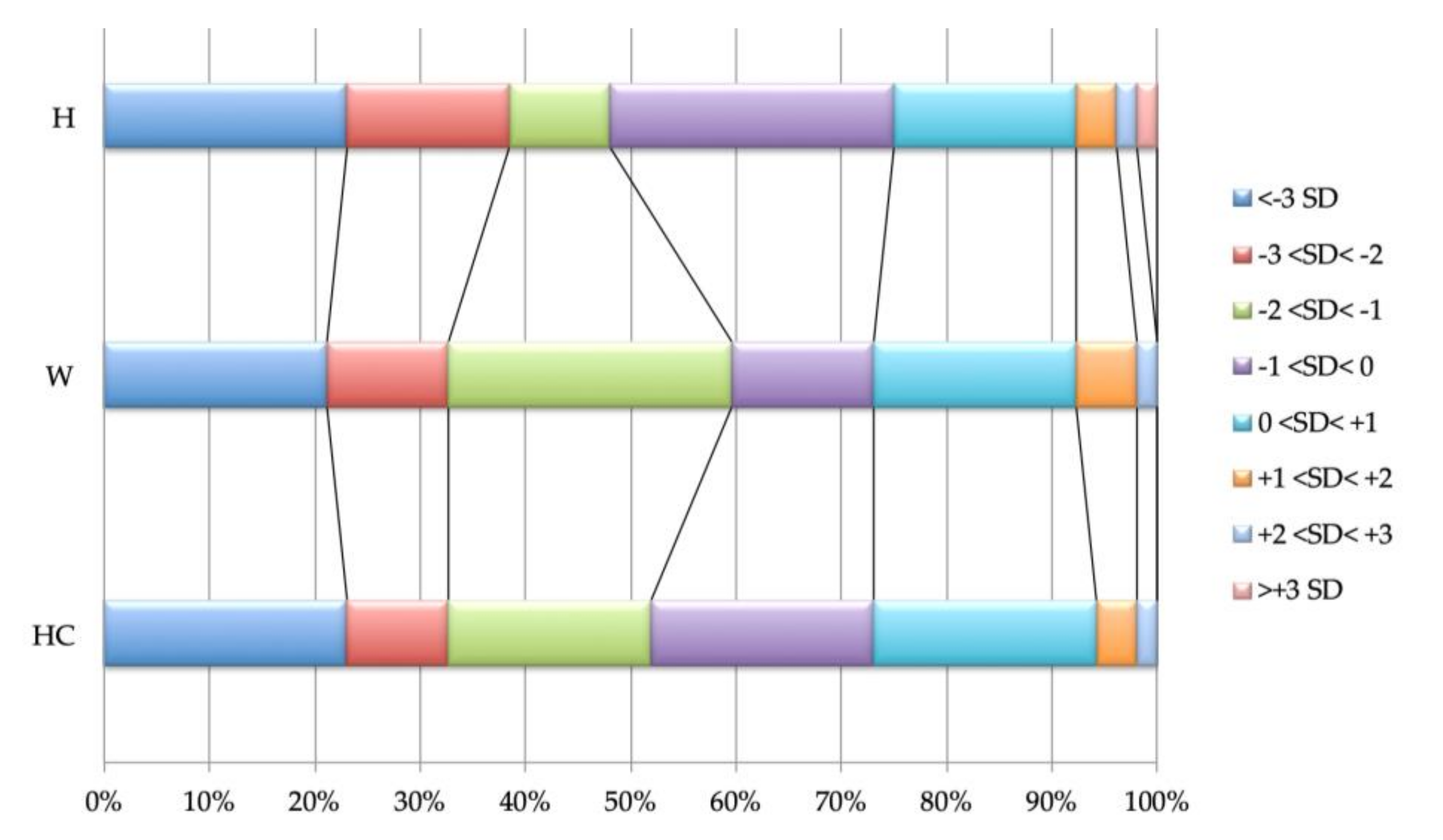
Growth parameters are shown in
Figure 2; most patients have poor growth, with approximately a third being under -2 Standard Deviations (SDs) for Height (H), Weight (W), and Head Circumference (HC); only 25% of the patients show parameters with positive SD values (≥0 SD). Microcephaly is present in 35% (18/52); in six patients, the head size reduction is proportional to their short stature, with a H/HC discrepancy lower than 1SD, while in 12 a more prominent slowdown of the HC is found. Absolute macrocephaly is never observed, but 21% (11/52) have relative macrocephaly. Height is below the normal age range in 35% (18/52) of the children, and in 61% (11/18) of the cases it is associated with poor weight; two children gained weight after a Percutaneous Endoscopic Gastrostomy (PEG) was performed (the QARS1 patient and one of the seven SCA subjects).
Facial dysmorphisms are present in 58% of children from almost all categories of disorders, except for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS), Friedreich Ataxia (FRDA), mitochondrial disorders, and NCL. Almost a third of children (16/52) present somatic malformations, which are in most cases skeletal deformities (75%), while heart and genito-urinary abnormalities are rare; eye malformations are limited to Gillespie syndrome, with 2/2 patients showing pathognomonic aniridia (see
Supplementary Material, Table S1 for details)
Consistently with the cerebellar dysfunction, a gait and/or appendicular ataxia is present in 80% of patients able to fulfill the examination tasks (35/44); in eight patients, the presence of ataxia is not assessable, given the severely compromised motor or developmental skills. A total of 27% of patients never acquired the first motor developmental stages (no head/trunk control). Tremor is present in 36% (16 of the 44 patients in which it is possible to explore the sign). Oculomotor abnormalities (apraxia or jerky smooth pursuit) are seen in 63% of the children (31/51, one patient has cortical blindness); this number may not reflect the real incidence, given the poor collaboration of the children. A total of 36% have strabismus (19/52) that is in 79% of cases convergent and in 21% divergent. Of the 36 children able to speak, more than a half have dysarthria (58%, 21/36). Impairment of the orobuccal motility (orobuccal dyspraxia) is present in 23% of patients, while two patients have dysphagia. All such signs do not appear to be specific to a particular disorder, but can be variably present—or not—in all the ataxic syndromes included in the cohort.
Hypotonia is seen in 56% (29/52) and in some cases, particularly in children with DNA repair disorders, a truncal hypotonia is associated with distal limbs spasticity. Extra-cerebellar neurological degeneration is common. 65% have signs of pyramidal system involvement (spasticity, hyper-reflexia, clonus, Babinski sign). Extra-pyramidal movement disorders are found in 21% and may be dys/hyperkinetic movements (four patients), choreoatetoid movements (three, all affected by DNA repair defect disorders: 2 AOA1, 1 A-T), dystonia (7 patients: 1 Cockayne, 1 Mitochondrial, 2 NBIA, 1 PCH, the QARS1 patient, 1 SCA); bradykinesia is observed in one patient with mitochondrial disorder. Finally, a hypo-reflexia is present in 15%; five out of eight of these children have a definite neuropathy recorded at NCS (one did not have the investigation, the Wolfram child and one Cockayne had normal NCS study).
Epilepsy affects 25% (14/52) children and in particular all children with NCL and PCH and 41% of those with DNA repair defect disorders.
Brain MRI was performed more than once in all but eight patients (85%); a progression of the atrophy is present in 70% (31/54). Children with the absence of radiological progression had ARSACS (2/2 children affected by such disease), CMT2Z (1), FRDA (1), Gillespie syndrome (1/2), KIF1A mutation (1/2), mitochondrial disease (two of the six with available follow-up), PCH (1/2 with available follow-up), SCA (2/5 with available follow-up, one SCA6, one SCA5), and Wolfram Syndrome (1/1). CA affects cerebellar vermis in all patients, while hemispheres are spared in seven cases. In 38% (20/52), a cerebellar hyperintensity is present, affecting dentate nuclei (7/20), the cerebellar cortex (7/20), or both (6/20). Cerebellar hypoplasia is present in six children (12%): 3/5 CDG and 3/3 PCH; in these latter three children, hypoplasia spreads to the pons, showing the pathognomonic PCH pattern. A brainstem involvement is globally present in 29%. Atrophy was present also in the supratentorial areas in nine cases; in two NCL and in two out of five Cockayne, it is associated with WM abnormalities, with these latter globally present in 33%. One child with Cockayne syndrome and one with NCL also have basal ganglia alterations.
In the majority of cases, neurophysiological studied have been conducted. All the children underwent at least one sleep-EEG, which demonstrated epileptic abnormalities not only in those patients suffering from epilepsy, but in an additional seven cases, so that the global prevalence of EEG abnormalities was 40% (21/52). BAEP and VEP were, respectively, pathological in 32% (15/47) and 20% (9/44) patients in which the exams were performed; both studies does not seem to correlate with any disorder; also, ERG alterations, present in 8 out of 47 patients (17%), are not typical of specific diseases, except for the fact that both CLN patients showed an abnormal retinal function. SEP study has been performed in a small number of patients (n = 19) addressing specific clinical suspicions (mostly, a possible SCA diagnosis); in such children, 58% showed abnormalities. EMG and NCS were, respectively, performed in 27 and 35 patients. EMG was pathological in 37% of the tested patients and NCS abnormalities were just as common (37%). Children presenting EMG + NCS alterations are seven (26%, 7/27), more than a half with DNA repair defect diseases.
Almost half of the patients underwent muscle biopsy. The exam showed non-specific alterations in 28%: minimal neurogenic alterations were present in three patients with DNA repair defects, while the exam revealed minor alterations in the structure of muscular fibers in four SCA patients. A definite single or multiple respiratory chain complex deficiency was found in 16%: all the four patients still lack molecular confirmation. A Coenzyme Q10 deficiency was present in two children, and in one of them the defect was confirmed by genetic analysis.
In the two patients with genetically defined NCL, lipopigments were present at the histological examination of skin biopsy.
Metabolic investigations has been changed and expanded over the years, so that not all children underwent the same tests. T-IEF was studied in 81% of children and was normal in all but the 4 patients with CDG, all presenting a type I pattern of glycosylation. investigations for DNA repair defect disorder (cholesterol, albumin and AFP dosage) has been also performed in 81% and revealed abnormalities in 14% (6/42) of patients; in all of them, the metabolic anomaly was consistent with the disease. TSH and Vitamin E were normal in all the patients tested. A total of 10% (4/42) patients had elevated levels of serum lactate and pyruvate. Serum AA and urinary OA profiles were studied in 85%, revealing AA alterations in one case and OA alterations in three cases: one child with Ataxia with Oculomotor Apraxia (AOA, who has AA + OA alterations), one metabolic disorder, and one mitochondrial disease (who both have excretion anomaly of multiple OA).
4. Discussion
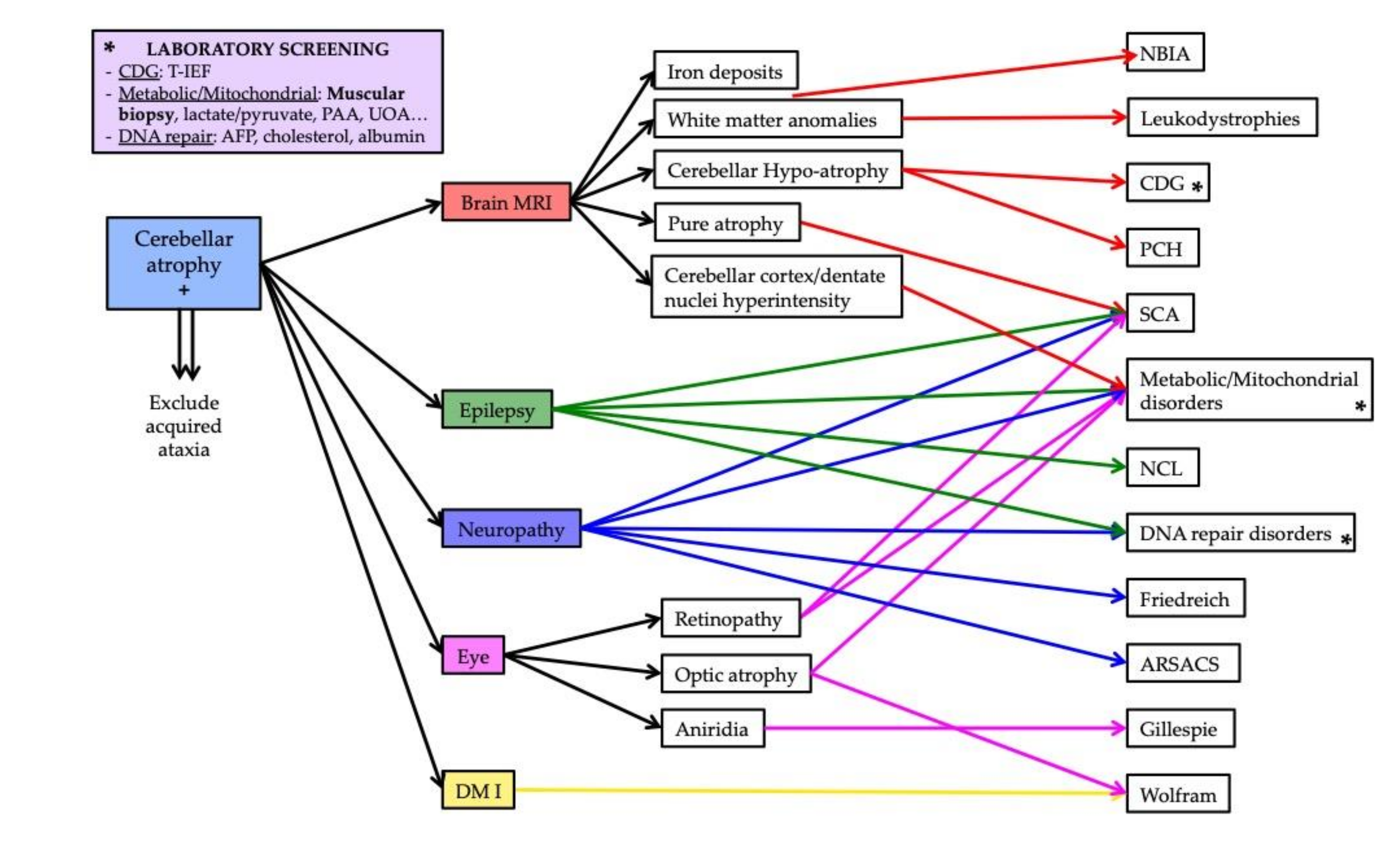
The aim of this study was to create a possible diagnostic workup for pediatric ataxias with cerebellar atrophy, which can guide both the formulation of a clinical suspicion and the interpretation of big genetic data; the proposed algorithm is shown in
Figure 3.
We do not include brain MRI as part of the diagnostic process as it should be the first exam to be performed in a child with a congenital or early onset ataxia [
2] in order to discriminate in the first place primary from acquired ataxias (e.g., cerebral tumors, toxins, teratogens, prenatal viral infections…), and, in the case of a primary ataxia, malformative to degenerative diseases. Studies have focused so far on delineating different neuroradiological patterns with CA, while little has been written on the global clinical presentation of the patients.
Neuroradiological evaluation is the prerogative and should precede any other investigation. In most patients, it was performed more than once and frequently revealed atrophy progression. Consistent with the literature data [
2], cerebellar vermis is always atrophic, but the degeneration is global (vermis plus hemispheres) in most kids, without any disease-specific pattern. Children who have a CA superimposed to cerebellar hypoplasia had those disorders in which cerebellar degeneration is known to start at a very early stage of development: PCH and CDG [
3,
11]. The hypo-atrophic pattern cannot be missed, as its presence consistently narrows the possible diagnosis to a small group of disorders. A hyperintensity of the cerebellar structure is otherwise less specific; it is generally thought of as a sign of
PLA2G6 mutations, mitochondrial diseases or, once again, CDG [
2,
8]. This is partially confirmed by our study, where more than a half of children with mitochondrial diseases or Neurodegeneration with Brain Iron Accumulation (NBIA) present dentate nuclei and/or cerebellar cortex hyperintensity, but the same sign is also shared by other kind of diseases (e.g., DNA repair disorders, SCA) as found in a previous work [
8]. This observation points out that the presence of dentate or cerebellar cortex hyperintensity alone is not sufficient to suggest a specific disease or disease group but, along with other features, it may strengthen the hypothesis of a metabolic disorder. Of note, the only PCH child with cortex and dentate T2-hyperintensity was affected by PCH6, caused by the mutation of a gene implicated in mitochondrial functions (
RARS2) [
12]. The involvement of brainstem structures is common and it is important to recognize the patterns that are disease-specific: beyond PCH, these include the characteristic pontine tigroid hypointensities of ARSACS [
6]. Brainstem atrophy is considered rare also in advanced stages
2; in our cohort, it is present in 9 out of 52 children.
Supratentorial alterations are mostly non-specific, as also found in previous studies [
8,
9], although, when associated with other features and consistent with the clinical phenotype, they may aid in the diagnosis (HLD in the case of diffuse hypomyelination, Cockayne syndrome with hypomyelination and calcifications, WM abnormalities and severe atrophy in NCL). Interestingly, of the three patients with mutation in NBIA genes, only one presents a definite basal ganglia anomaly until now.
Clinical presentation is often similar in all children with CA. Patients who are sooner brought to the physician attention are mostly affected by diseases with intrinsic malformative aspects (e.g., aniridia in Gillespie) or characterized by an early cerebellar dysfunction (e.g., CDG or PCH). Ataxia or other signs of cerebellar dysfunction may not be present at onset and emerge over time; most children who reach sufficient motor skills develop truncal or appendicular ataxia, as well as oculomotor functions are impaired in the majority of cases. Oculomotor abnormalities and orobuccal dyspraxia are common but not disease-specific, although strabismus is almost uniformly found in CDG. Muscular hypotonia affects more than a half of children at any age and may be the sign of a cerebellar and early pyramidal involvement; in younger children, the sign may disappear with age and evolve into ataxia or, in some cases, turn into spasticity. Our clinical data confirm that a combined pyramidal and extrapyramidal dysfunction is a clue of DNA repair disorders [
7]. This latter disease group is also the one most frequently complicated by epilepsy, suggesting that DNA repair disorders must always be considered in children with ataxia, epilepsy, and microcephaly (also a recurrent finding).
Unless we have a definite suspicion based on brain MRI and clinical evaluation, we need the help of neurophysiology and laboratory tests to narrow the field of differential diagnosis.
Abnormalities in EEG are often found in children with mitochondrial/metabolic disorders, even in the absence of seizures, as an effect of global metabolic dysfunction on brain activity. Alterations in ERG are generally considered a sign of mitochondrial diseases or some specific SCA, mainly SCA7 [
9,
10,
11,
12,
13], but they do not seem to recur in particular groups of diseases in our cohort, and the same happens for visual and auditory evoked potentials. VEP may give a strong clue to diagnosis, for example revealing signs of optic nerve involvement in Wolfram Syndrome. In such studies though, the poor collaboration of younger and severely disabled children must be considered. SEP appears helpful, showing a dysfunction in the somatosensory system functioning, at different levels, in those disorders known to possibly have such features (ARSACS, DNA repair defects, FRDA, NBIA, Wolfram syndrome); unexpectedly, neither of the two SCA children who underwent the exam had SEP abnormalities.
PNS involvement sometimes complicates the clinical presentation and course of children. In our cohort, EMG and NCS data were helpful to guide genetic analyses, in some cases focusing on a very specific disorder. Patients with NCS abnormalities were affected by ARSACS, Charcot–Marie–Tooth 2Z (CMT2Z), DNA repair disorders, FRDA, and mitochondrial diseases; EMG was altered in the same disorders, plus NBIA and PCH. The highest recurrence of PNS involvement was found in children with DNA repair defects: this demonstrates that, even if EMG is quite an invasive exam to perform in a child, it must be considered as it may lead to diagnosis. Notably, the
PNKP gene was recently described to cause a large spectrum of phenotypes, ranging from congenital macrocephaly to adult-onset ataxia [
14]; we suggest that in children where ataxia is associated with at least two of the following three features—microcephaly, seizures, and neuropathy—
PNKP sequencing should be considered, especially in the presence of supportive elements such as AFP/cholesterol/albumin alterations or signs of extrapyramidal system involvement.
The role of muscle biopsy in the diagnostic process of pediatric ataxias is already recognized and biochemical studies on muscle samples were found to be helpful in a range from 5.5% up to 23% of children [
8,
15,
16,
17]. In our cohort, a definite alteration was found in 24% of children, thus confirming the role of the exam in the diagnostic process of pediatric-onset CA.
Lastly, the study proves the effectiveness of a laboratory investigations for conditions that may present with CA in the phenotype (and therefore genotype) definition (results summarized in
Table A3). Abnormalities of serum AA, urinary AO, T-IEF, cholesterol, albumin, and AFP were found in a proportion of patients and in such cases unambiguously defined the disease group. It is important to remark that such exams may be repeated in children who lack diagnosis, as they can turn positive over time: in one of the two CDG brothers of the study, T-IEF was negative at onset (10 months), but became positive at the age of 6 years; this result allowed us to give a diagnosis in two children in which WES failed to find the molecular defect (as only one mutation in the CDG1L gene could be detected). Of note, negative results in muscle biopsy or normal serum lactate and pyruvate values should not be considered exclusion criteria of mitochondrial-based pathology, as demonstrated by the PCH6 patient, who both had values in the normal range, despite PCH6 being the only PCH with mitochondrial involvement. Mitochondrial diseases are among the most common cause of degenerative ataxia in children [
16]. This is confirmed by our study, where the leading cause was DNA repair defect disorders, followed by mitochondrial diseases and SCA. It is therefore important to exclude all such conditions, also considering that pediatric SCA often does not show a nucleotide expansion pathogenic mechanism. Of importance, the Array-CGH was normal, confirming the poor role of cytogenetic in pediatric ataxias [
18].
In conclusion, this study proposes a battery of investigations that it is useful to perform when approaching a child with cerebellar degeneration, based on a retrospective evaluation of a cohort. Beyond brain MRI, and especially in those patients who already underwent molecular analyses without a successful diagnosis, we support the use of neurophysiology and muscular biopsy, as they can at least allow a biochemical confirmation and narrow the field of genetic diagnosis. Additionally, we encourage performing metabolic investigations, as they are rapid and cost-effective and, when positive, they can be conclusive in resolving the diagnosis.


 ,
, 



{kind=link}
{kind=link}
{kind=link}


