4.1. Uranium Speciation in Groundwater
In the study samples from wells near the sludge repository, uranium at concentrations of up to 1.56 mg/L was found (
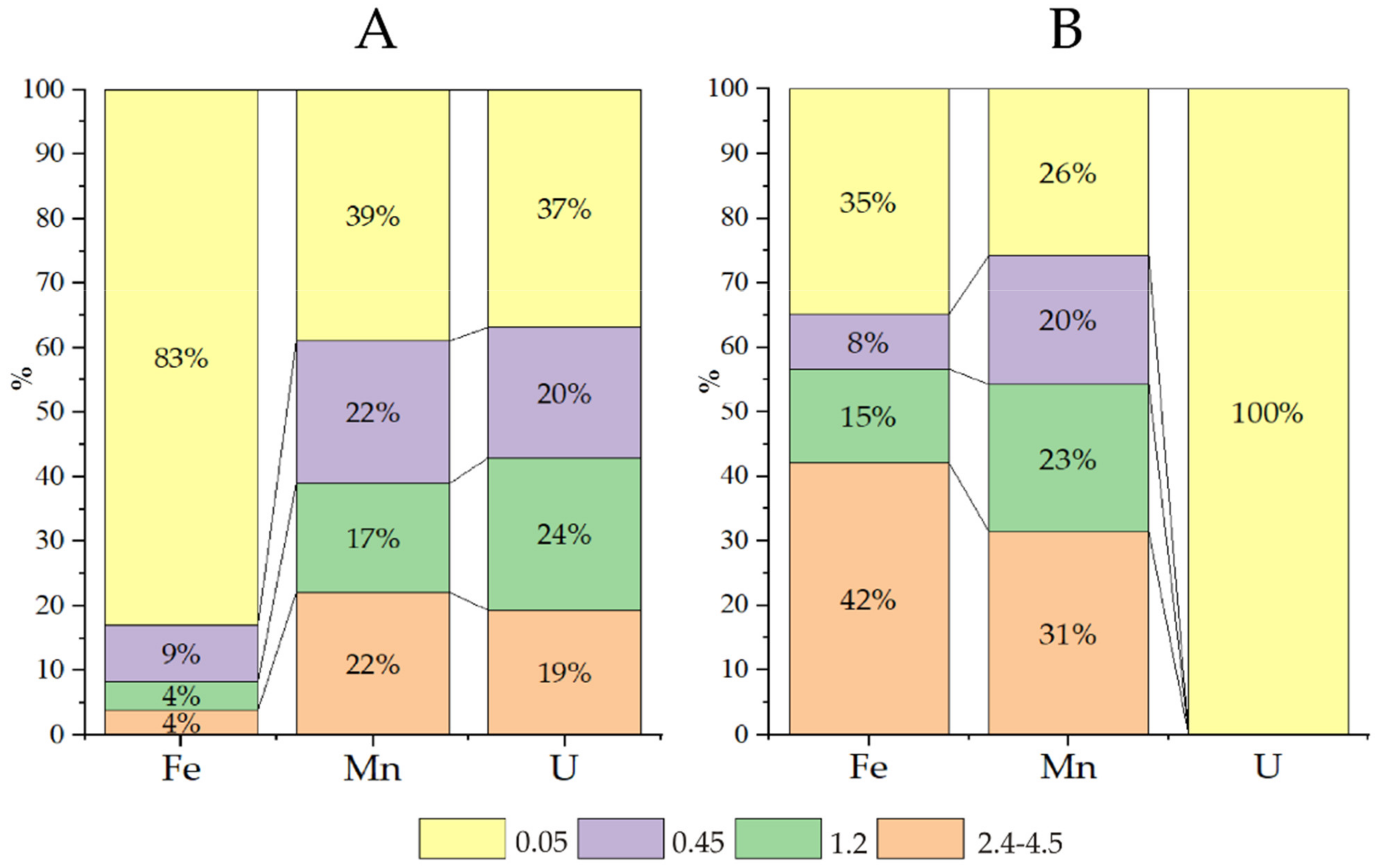
Table 1). Cascade filtration of the water samples from Wells 2 and 3 in an anaerobic box under field conditions allowed us to establish the size distribution of the main particles associated with uranium migration. Chemical elements in groundwater can migrate as suspended particles (0.1 μm), colloids (1–100 nm), and molecular solutions (≤1 nm). It is generally accepted that suspended particles are nearly nonexistent in groundwater, colloids play a subordinate role, and truly dissolved forms dominate. Exceptions are elements such as Si, Al, Mn, Fe, U, and Th. We therefore studied their behaviour (
Figure 1).
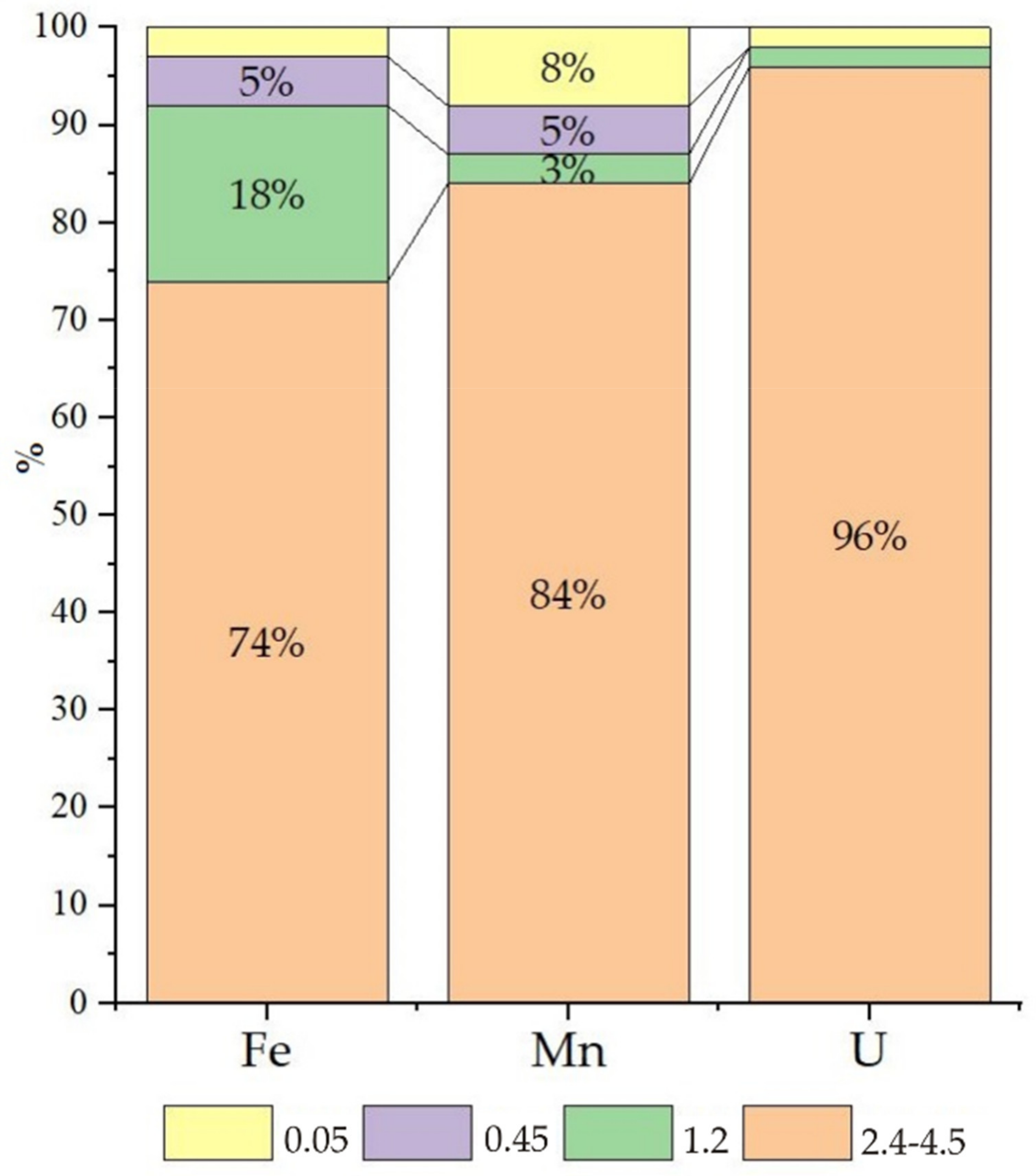
More than 6 g/L of nitrate ions was found in the water of Sample 3, contributing to slightly oxidative conditions (Eh 90 mV), and uranium was found mainly in the form of uranyl ion carbonate complexes, in which form uranium easily migrates (
Figure 2 fraction < 0.05 μm, yellow colour). Iron and manganese are transported mainly as colloids, and the large fraction (2.4–4.5 μm) accounted for 30–40% of these elements. For iron, this fractionation is explained by the formation of insoluble Fe(III) (hydro)oxide; the explanation for manganese is not as apparent.
According to the calculations (
Table 2), manganese should be present in the form of Mn
2+ and MnSO
4(aq) without solid phases. Moreover, the aerobic oxidation of bivalent compounds to insoluble forms with the highest valence, followed by subsequent separation, is used to precipitate manganese from contaminated water. Currently, we can only assume that the concentrated Mn colloids are organomineral compounds.
The water of Sample 2 presents a nitrate content of approximately 1 g/L (Eh −28 mV). Iron is mainly in the dissolved forms of Fe(II)—Fe2+, FeHCO3+ and FeSO4(aq)—while manganese is in the solution (40%) and in all fractions of colloids. Note that in this water, the concentration of manganese (2.3 mg/L) is much higher than the MAC. According to the calculations, in zone 2 at a distance of tens of metres from the sludge pond, the uranium in solution is mainly in the form of U(OH)4(aq) and in the form of true and pseudocolloidal UO2(am).
Nitrates, with which the solutions of Samples 2 and 3 are saturated, form oxidative conditions in neutral groundwater in the territory of the NCCP and prevent the removal of uranium. Moreover, a positive shift is evident even when the NO
3− content decreases from 6169 mg/L to 1124 mg/L: (a) the oxidation state of uranium changes from U(VI) to U(IV), and (b) the content of ferrous iron (Fe(II)), a potential electron donor, increases from 0.01 to 10.7 mg/L. According to calculations and a review [
26], for the intense reduction of nitrates at pH 7, Eh ≤ 0 mV is necessary. In our case, such values are characteristic only for Sample 1 (background water). Even in this case, at an Eh of -190 mV, sulphate is predominant over sulphide species in the solution (
Table 2), with values of 3.8 × 10
−5 H
2S
total and 2.74 × 10
−4 SO
42− total. Modelling experiments of the metabolism of biota were therefore designed to attain the reduction of nitrates and sulphates during NCCP groundwater treatment. The formation of sulphide will lead to the removal of heavy metals, and sometimes this mechanism is the only opportunity for the remediation of certain pollutants, such as zinc and other heavy metals.
4.3. Laboratory Modelling of Biological Groundwater Treatment
In the first stage, electron donors were selected to stimulate the microbial community of the formation water in Samples 1 and 2 (
Table 4). Ordinary substrates and one complex substrate (whey) [
36] were used. It has been established that the optimal electron donor for the denitrifying microbial community is whey, a rich substrate containing a variety of organic and mineral substances, including phosphate [
37]. Further, whey was used in the in situ experiments.
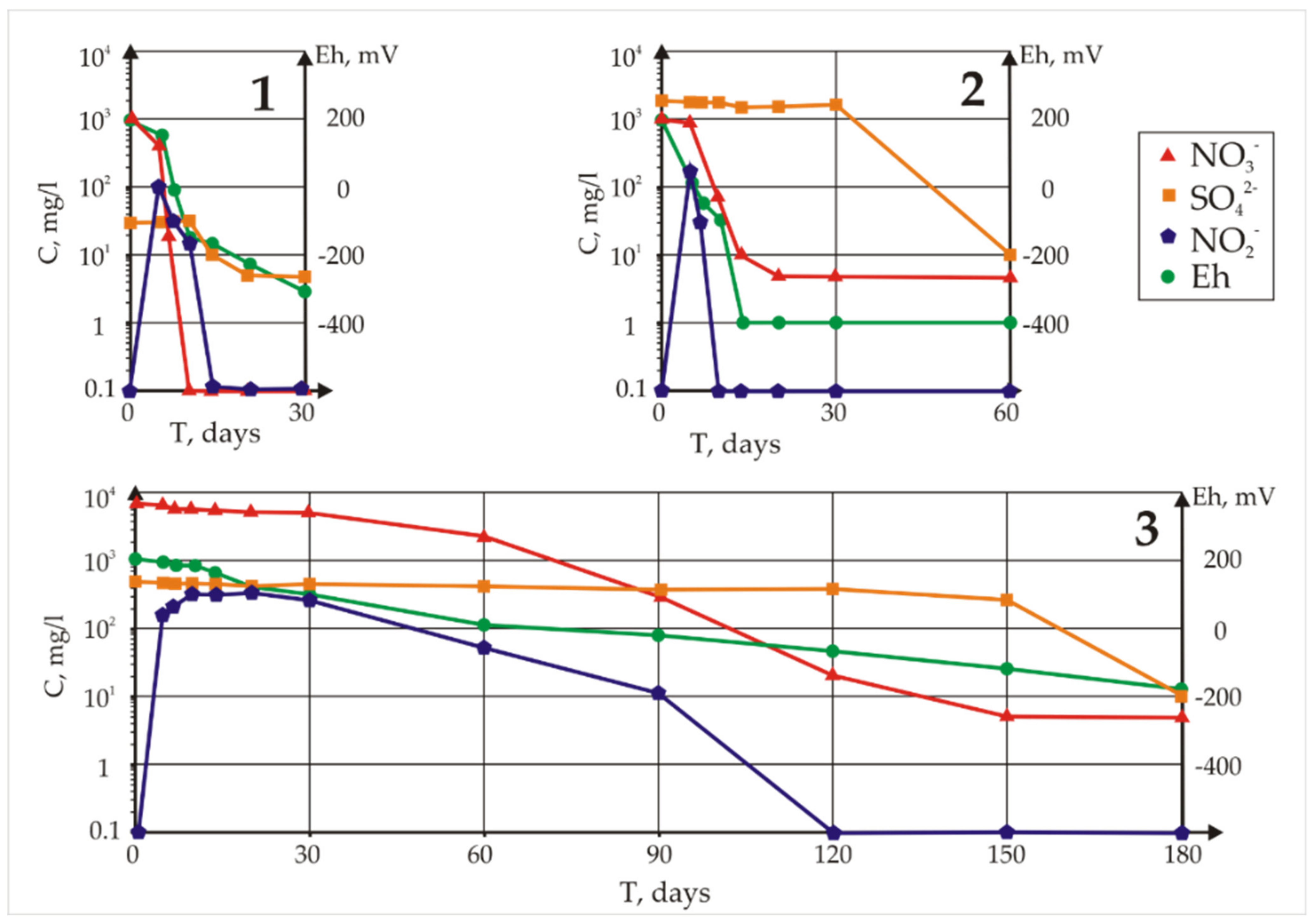
The measured concentrations of sulphates, nitrates, and nitrites and the Eh values in the samples with whey are given in
Figure 3. First, a decrease in the concentration of nitrate ions took place, and only after their complete removal did the reduction of sulphate anions begin. In Sample 1 from the background well, where 1 g/L of nitrate was specifically added, the process of nitrate reduction lasted approximately 7 days, and the visible removal of sulphates began after 20 days. In Sample 2, nitrate removal and sulphate removal continued for 20 days and 60 days, respectively. In the sample from Well 3 with maximum pollution, nitrate removal continued for 150 days, while a significant accumulation of nitrite was observed. Removal of sulphates lasted 180 days. Thus, in natural water with the original microbial community, purification is very rapid. It should be taken into account that the initial mineralization and redox potential of the background water (Sample 1) were significantly lower, with only 30 mg/L sulphate.
To explain the fundamental differences between the nitrate and sulphate removal rates in the laboratory experiments, 16S rRNA gene analysis of the microbial community was conducted at the end of the whey addition experiment (
Table 5).
Whey addition to Sample 3 from a highly polluted site led to a significant decrease in microbial diversity and the dominance of the genera
Rhodococcus and
Rhodobacter. The former is known for its ability to perform aerobic denitrification; it grows on media with a complex source of organic matter, such as media containing sugar beet molasses and sucrose as carbon sources [
38].
Rhodobacter can reduce nitrate and nitrite ions under anaerobic conditions. Some members of the genus Bacillus reduce nitrates and sulphates and digest rich organic substrates.
In the development of the microbial community of Sample 1, after whey addition, representatives from the taxa Acidovorax, Hydrogenophaga, and Thauera dominated; these microorganisms are known for their ability to reduce nitrates, iron, and other compounds. The presence of sulphur cycle bacteria—representatives of the genera Sulfuritalea, Desulfobacterium, Desulfomicrobium, and Desulfuromonas—was also observed.
The removal of nitrates and sulphates in Sample 1 thus occurred after the addition of whey due to the activity of a metabolically diverse microbial community represented by denitrifying and sulphate-reducing bacteria. For highly contaminated Sample 3, nitrate removal was too slow because of the activation of nonspecific nitrate reducers. Such retardation can be explained by toxic nitrite shock, which lasts for a long time and occurs at the initial stage of the denitrification process. For nonspecific denitrification bacteria of the genera Rhodococcus and Rhodobacter, nitrate removal was insufficient; however, a decrease in the rate of NO3− reduction led to lower NO2− concentrations to avoid nitrite shock. We stress again that not only the amount of nitrates but also the microbial community diversity causes retardation in the nitrate reduction process.
It should be added that in conditions of high pollution, the development of microbial biofilms on rocks can play a key role in contributing to pollutant concentration gradients and the protection of cells from toxic stress [
39]. Microbial biofilm development can likely play an important role in formation conditions with high contamination. Therefore, we studied this process in detail.
4.4. Assessment of Biofilm Formation and the Role of Biofilms in Uranium Immobilization
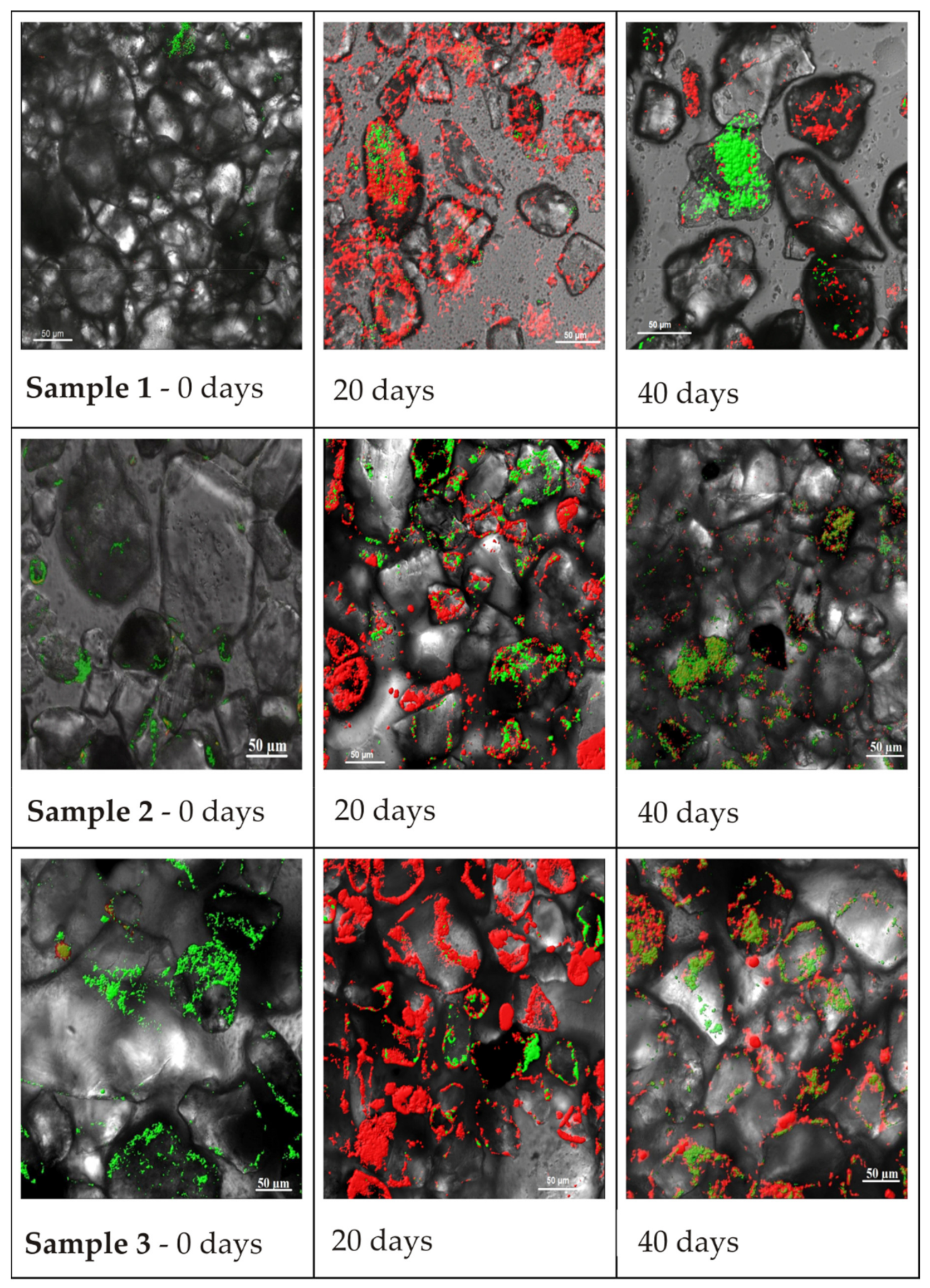
In laboratory experiments using several methods, the formation of microbial biofilms by microorganisms on loam (sandy loam) from boreholes 1, 2, and 3 after single glucose-acetate activation was investigated. Microphotographs obtained using confocal microscopy (
Figure 4) show maximum biofilm development on the 20th day of the experiment. On the 40th day, the biofilm matrices of microbial communities on the three rock samples deteriorated, and only in some areas of the rock did attached microorganisms remain.
Based on visual analysis, the maximum accumulation of cellular nucleic acids (green colour) before glucose activation was observed in the loam of Sample 3. The maximum accumulation of polysaccharides on the 20th day was observed for the rock of Sample 1, but on the 40th day, the maximum polysaccharide amount was observed for the loam of Sample 3, indicating uneven biofilm development on rocks during interactions with solutions of different compositions. On the rock sample suffering from maximum technogenic impact, biofilm development was slower on the 20th day, but the deterioration rate on the 40th day was also not rapid.
The confocal microscopy visualization data were confirmed by the MTT assay data (
Table 6). The respiratory metabolism of microbial biofilms reached the maximum on the 20th day, with the largest respiratory activity observed for the microbial community of Sample 1, which was in contact with groundwater and the minimum observed for the rock of Sample 3, which was mostly affected by nitrate sulphate water.
The maximum area occupied by biofilms before microbial stimulation was largest on the sample with maximum contamination, and after stimulation, the area was largest on the uncontaminated sample. It should be noted that on the 40th day, the biofilm area on Sample 3 was two times larger (15.5%) than the areas measured on the other samples. The formation of a more developed biofilm and the lower rate of biofilm deterioration on the most contaminated rock may indicate the important role of biofilms in microbial processes with a high nitrate background.
It should be added that, based on the analysis of 16S rRNA genes in the biofilms, on both samples the dominance of representatives of the families Comamonadaceae (genus Acidovorax) and Pseudomonadaceae (genus Pseudomonas) was observed.
The formation of biofilms on rocks led to a change in the sorption capacity of the samples towards uranium. Samples after 20 days of the experiment were used as sorbents (
Figure 4,
Table 6). The K
d values calculated from the experimental results are presented in
Table 7. A notable increase in the K
d value occurred for the rock of Sample 3, with increases of 24% and 30.5% (for distillate and groundwater solutions, respectively), and for Sample 1, with values of 33% and 37%, caused by the coating of particles with biofilms. We associated the higher sorption characteristics for Sample 1 with the higher degree of biofilm coating of the particles, explained by an increase in the number of sorption sites on the polysaccharide matrix on the rock surface, including carboxyl and hydroxyl groups [
40].
4.6. Estimation of Biogeochemical Mineral Neoprecipitation
Two precipitate generation phenomena were recorded in the experiments after whey addition. The first (bright coloured) precipitate appeared a few hours after cultivation. The second (dark coloured) precipitate originated after the reduction of sulphate ions for 1–6 months. Scanning electron microscopy (SEM) solid-phase analysis revealed two dominant phases differing in both morphology and particle composition. The first phase (
Figure 6A) is represented by heterogeneous particles with abundant sharp faces; the appearance of this phase in the first hours after mixing the solutions suggests that the disequilibrium of the solution arising from mixing is the source of its formation. The phase contains high concentrations of phosphorus and calcium without uranium. The second phase is a sintered aggregate containing a large number of spherical and elongated particles of 1–5 μm (
Figure 6B). This phase contains considerably more S, Fe, Na, and K, with high concentrations of uranium of up to 5.36 wt. % (
Table 8). According to the chemical composition, we may assume that sulphides were added to the first (apatite-like) phase over time, as evidenced by the sharp drop in the concentration of sulphate in the solution (
Figure 3).
Based on the data obtained, it can be concluded that nitrate reduction can be successfully activated by the addition of organics to groundwater aquifers in situ. A decrease in nitrate concentration reduces the migration activity of uranium since the associated decrease in Eh leads to uranium reduction and immobilization. The risk-free prediction of uranium behaviour and risk assessment of uranium remobilization remain important tasks. There are a group of factors that determine the behaviour of uranium in biochemical processes and have different influences on its (im)mobilization. These factors include the following:
- -
Microbial catabolic reduction of uranium, leading to the formation of an amorphous uranium phase (“biogenic uraninite”), which has a much higher solubility than other forms of uranium (uraninite logKs −4.84; UO2(am) logKs 0.11) and is very important for the design of more effective posttreatment reduction strategies.
- -
Uranium sorption on rocks that can be covered by biofilms of different compositions and intensities. The effective sorption sites of rocks and the formation of new bioorganic bonding sites in the polysaccharide matrix of biofilms may overlap. In our case, the Kd value of uranium increased by 30.5% in the most polluted well (6169 mg/L NO3−).
- -
Changes in Eh and pH values, affecting the distribution of uranium forms, the solubility of mineral phases, including clay phases, and the bacterial community itself. Abiogenic and biogenic reduction processes contribute to Eh reduction and pH growth, which are sometimes above the optimal values for denitrification bacteria (2–4):
- -
The formation of carbonate species in solution and carbonate mineral phases during the oxidation of organics (2–4) can enhance the mobility of uranium due to stable carbonate complexes or modify the sorption properties of host rocks.
- -
The formation of microbial metabolites that, on the one hand, inhibit nuclide migration when accumulating in biofilms and, on the other hand, enhance migration in the form of biocolloids (pseudocolloids), which mobilize uranium.
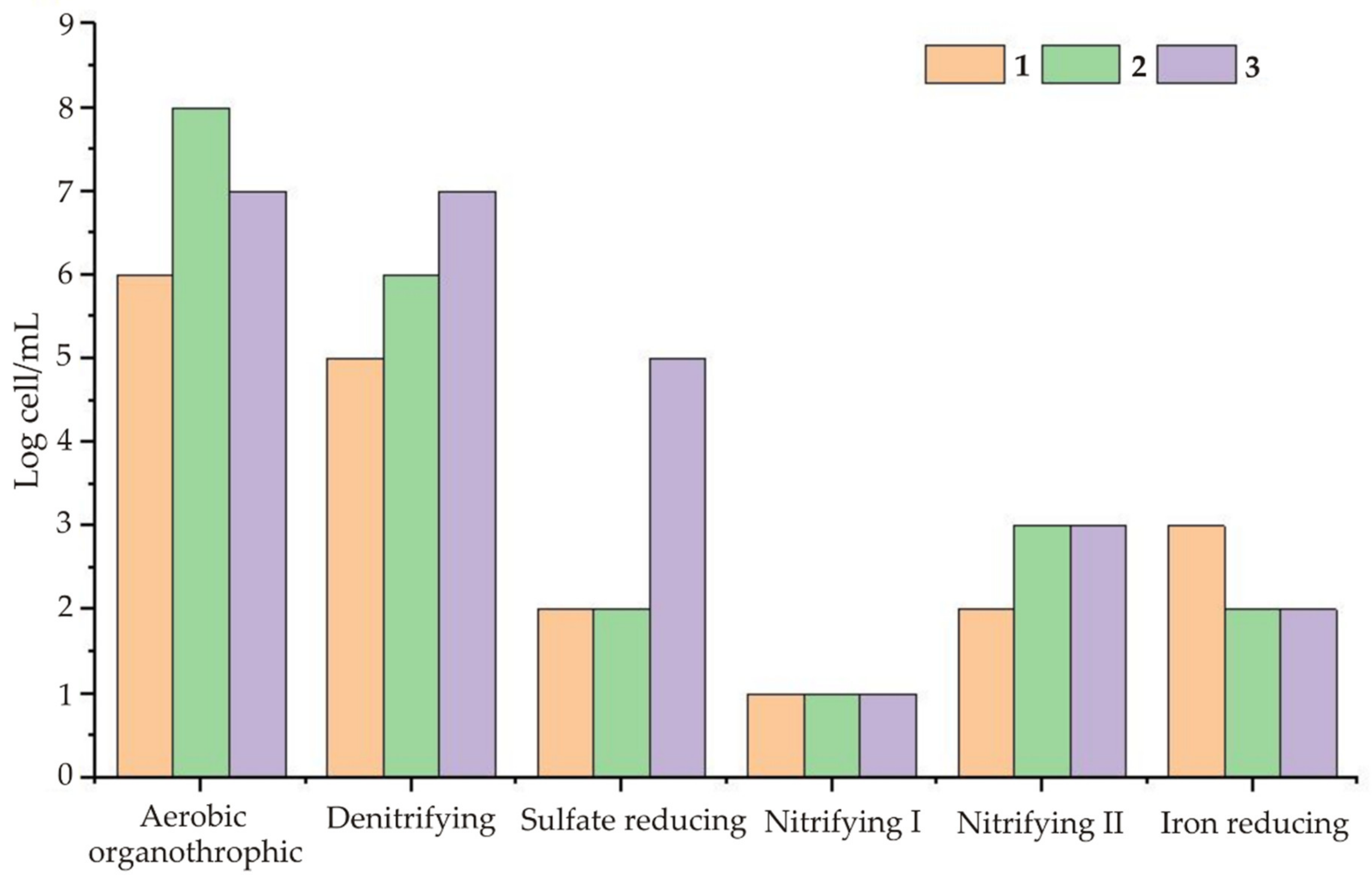
In addition to the above factors, the hydrogeochemical conditions at each facility play a crucial role. The maximum number of denitrifying and sulphate-reducing bacteria (
Table 2) was found in Sample 3, which was taken from the most contaminated well. Moreover, uranium, like many other metals, can be used in enzymatic reactions as an oxidizer. The question is, at what concentration does nitrate lead to a decrease in the biological efficiency of uranium reduction? This question should be solved experimentally. In particular, in the laboratory experiments, after the addition of 50, 500, and 3000 mg/L of NO
3− and 50, 100, and 200 mg/L of uranium to clean groundwater (Sample 1,
Table 1), we observed that at nitrate concentrations of up to 500 mg/L, the microbial reduction efficiency of nitrate ions increased within 7 days. At high concentrations of NO
3− and 200 mg/L of U(VI), pronounced retardation was recorded, up to 1.2%.
Microbial biofilm formation was studied under simulated underground aquifer conditions, and the maximum formation was observed on the 20th day. At the beginning of the experiment, the maximum intensity of respiratory metabolism of the attached microorganisms and the maximum coating area were observed on the rock from the contaminated well. The additional evidence that on the 40th day, the biofilm deterioration rate was lower on the rock from the highly contaminated sample than on rocks from other samples also indicates the importance and stability of the attached forms of microorganisms in conditions of high pollution. Since polysaccharides are the main component of biofilms, sometimes occupying up to 95% of the total area of the biofilm, their contribution to the immobilization of uranyl should be treated as the most vital. The surface of the polysaccharide matrix contains a wide variety of functional groups, primarily hydroxyl, carboxyl, carbonyl, and sometimes amino groups. In one of our publications, we performed a detailed analysis of the interaction of uranyl ions with the functional groups of o-polysaccharides isolated from the bacterium Pseudomonas veronii, a common biofilm inhabitant. Carbonyl groups have been found to play a major role at neutral pH values [
41]. Kazy et al. [
42] provided evidence of uranium bonding with the extracellular polysaccharides (EPS) produced by
Pseudomonas aeruginosa strain BU2 involving carboxylic groups at pH 5.0, whereas at lower pH values, amino and hydroxyl groups played a major role. It is worth noting that biofilms are a condition for the immobilization of other compounds, primarily iron, calcium, and chromium, that contribute to the formation of new mineral phases in biofilms (calcites, ferrihydrites, etc.) [
43] that are known for their high sorption capacity for many actinides [
44]. Taking into account the possibility of stimulating microbial processes, microbial growth on rocks can therefore be an important factor in the immobilization of uranium in the form of organic and organomineral deposits in local areas.
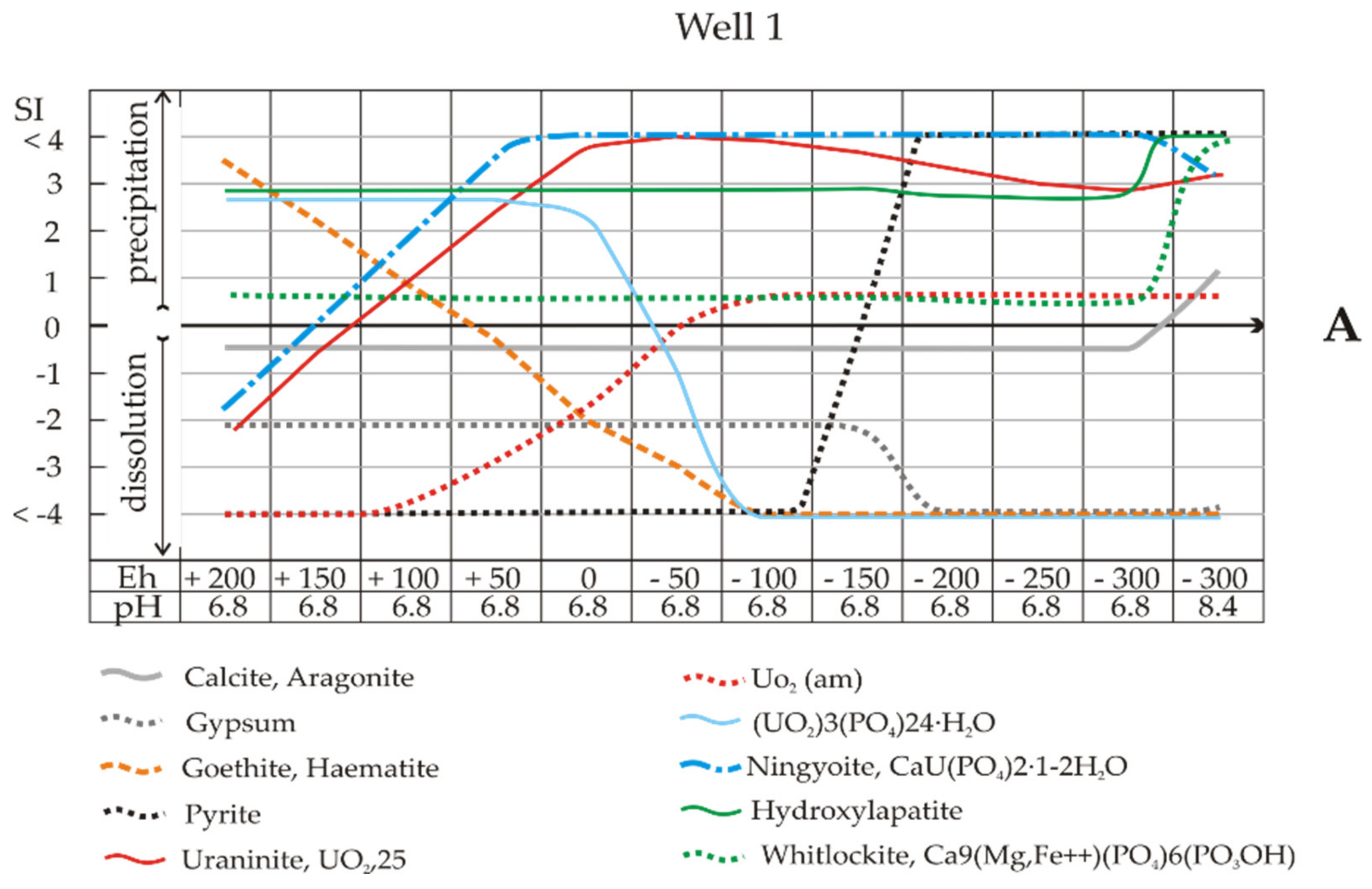
Calculating the SIs of solutions with respect to certain minerals in a wide range of redox conditions is important for the development of remediation practices since SI reveals how microbial activity changes Eh (
Figure 3). Recall that in the model calculations, 10 mg/L of uranium was added, corresponding to the simulated, not natural, solutions. Phosphates appeared due to the addition of whey to stimulate biota development. Thermodynamic modelling of the element speciation in sampled in situ solutions allowed us to track the change in valence of a number of elements at different Eh values (
Table 2). It should be added that the main calculation was performed at a fixed pH (e.g., 6.8). Microbial denitrification is known to be followed by an increase in pH [
45,
46]. In our experiments, no visible change in pH was observed; however, such a process cannot be completely excluded with a high content of nitrate. In the calculations, conditions with more alkaline solution values were therefore simulated to show the direction of the possible evolution of the system when the pH changed.
At pH 6.8 and Eh −100 mV (Well 1 and Sample 1), the mobility of uranium can be limited by the deposition of UO
2(am) (
Figure 7A), but these are the boundary pH-Eh conditions. Even a small amount of oxygen access (shifting to the left along the X-axis) will lead to the failure of pyrite deposition due to the lack of sulphide (black dots) and the preservation of uranium in solution. Note that at the same critical point, the solutions are completely undersaturated towards iron hydroxides (brown dotted line) and uranium phosphate (UO
2)
3(PO
4)
2·2-4H
2O (blue line). Natural background water is undersaturated with respect to calcite and biomineral aragonite at up to Eh −300 mV (pH 6.8), and the water will thus be vulnerable to acid drainage. Leaching of the system will result in the supersaturation of the solution with hydrocarbonate ions and calcite precipitation.
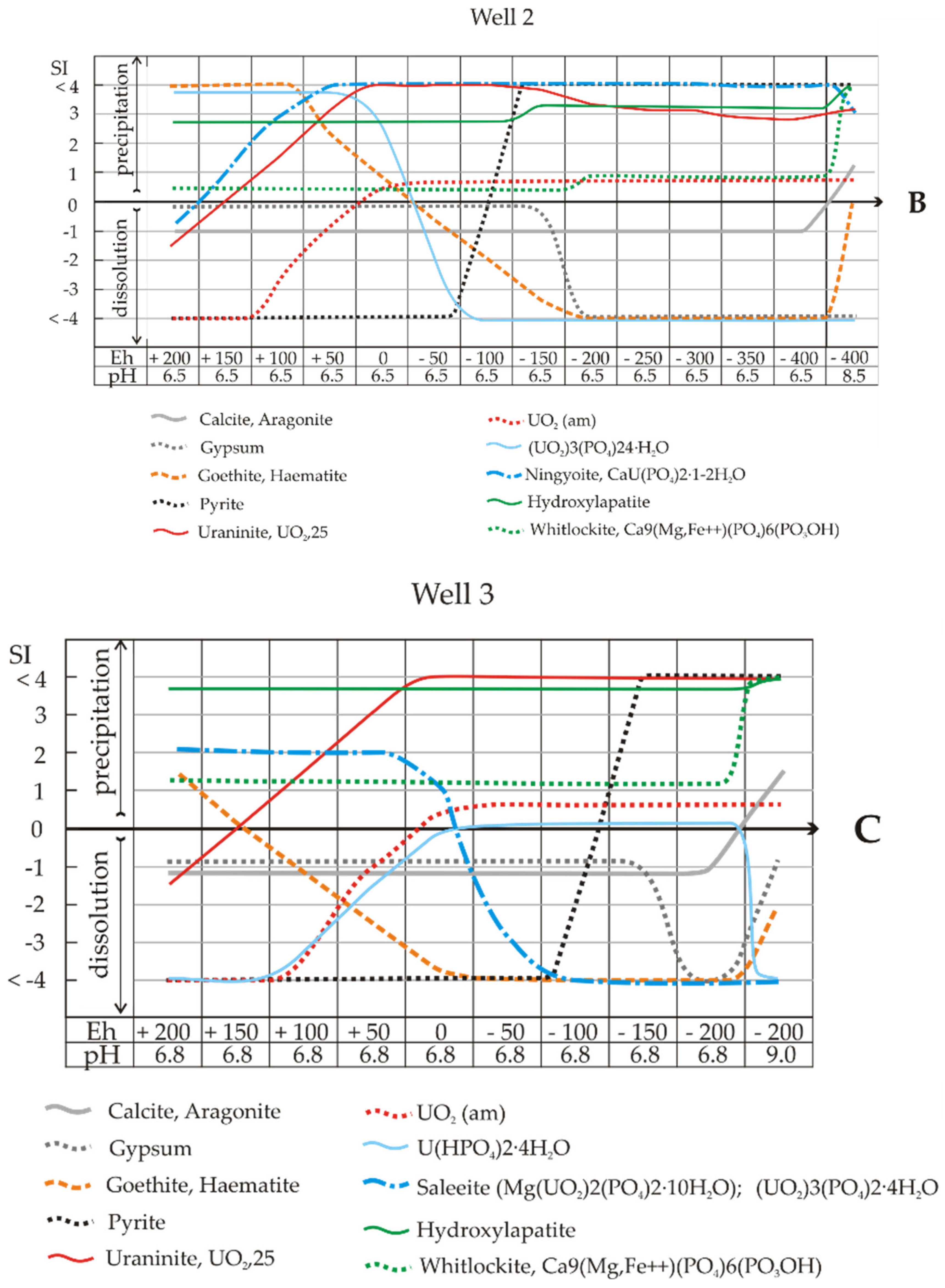
In Sample 2 (Well 2), the processes of nitrate and sulphate removal were quite slow in the experiment (complete reduction on the 20th and 60th days, respectively). Nevertheless, we observed an Eh value of −400 mV on the 15th day of the experiment. Thus, according to the model calculations, under biota stimulation in Sample 2, the removal of uranium from solution’n may occur primarily due to the precipitation of (UO
2)
3(PO
4)
2·2-4H
2O phosphates and subsequently of U(IV) oxides. The SI lines of these chemical species intersect at 0/−50 mV. This means that on the left side of the graph (Eh > −50 mV), phosphates should be precipitated from the solution, and a white precipitate should be formed, while at Eh < −50 mV, a black precipitate should be formed because of pyrite deposition (as in the experiment). However, the uranium content in the white precipitate is below the detection limit;, i.e., these are common calcium phosphates (see
Table 7). As in the previous case, leaching of the system will cause the solution to be supersaturated with hydrocarbonate ions and calcite precipitation. Under these conditions, goethite and hydrogoethite reach equilibrium, and their dissolution ceases.
A recent article on uranium phosphates demonstrated that uranyl–phosphate minerals can maintain extremely low levels of aqueous uranium in groundwater due to their low solubility [
47]. Once formed, such minerals are relatively insensitive to the system redox potential and are also more resistant to dissolution than other uranium minerals under oxic conditions outside of their stability fields [
48]. However, the pH range and phosphate-to-bicarbonate ratio need to be precisely adjusted to the correct values to achieve phosphate formation, and the exact conditions depend on the uranium and phosphorous starting concentrations. Future experimental work should be performed to understand the connections between solution chemistry and uranium mineral composition under our conditions (Samples 1–3).
The solutions were undersaturated with respect to calcite (SI = −1), although they were in equilibrium with gypsum to Eh -200 mV, i.e., at the beginning of the intensive bacterial reduction of sulphate. As shown above (reactions 2–4), during bacterial reduction, proton uptake reactions occur, and the pH increases. On the right side of
Figure 7A,B, at an alkaline pH of 8.5, the supersaturation of the solution towards biogenic carbonates is seen. The influence of uranyl speciation and iron oxides on uranium biogeochemical redox reactions was discussed previously [
49]. The presence of calcium in carbonate-bearing solutions promotes the formation of ternary complexes, Ca
2UO
2(CO
3)
3 and CaUO
2(CO
3)
32−, which have a profound impact on U biogeochemistry by decreasing both microbial and abiotic reduction rates. This is clear evidence; moreover, note that at pH 8.5 and Eh -400 mV, there is a peak in the formation of goethite or haematite corresponding to a sharp extension of their stability fields in the Eh-pH plots (
Table 8). According to the cited authors, “goethite and haematite act as sorbents of Ca and, as a result, decrease the proportion of less-reduced Ca
2UO
2(CO
3)
3 species, thus increasing the rate of microbial reduction of uranium compared to that in systems with no ferrihydrite”.
The calculation of SIs showed oversaturation of the solution of Sample 2 with respect to ningyoite–phosphate U(IV) at all Eh values (
Figure 7B). Ningyoite is a rare mineral; however, U(IV)–phosphate minerals may play an important role in U retention in mining-contaminated wetlands and as possible products of microbial U(VI) reduction [
50].
In Sample 3, from the well with maximum pollution, the composition of mineral associations during the Eh decrease changed considerably compared to the results in
Figure 7A,B. First, in the experiment, even on the 180th day, an Eh of only -200 mV was achieved. Second, ningyoite CaU(PO
4)
2·1-2H
2O dissolved, which corresponded to a high and permanent supersaturation of the pure solutions. Instead of ningyoite, U(VI) phosphates appeared—saleeite Mg(UO
2)
2(PO
4)
2·10H
2O or (UO
2)
3(PO
4)
2·4H
2O. These solutions are generally in close equilibrium with UO
2(am) until strong alkaline conditions, which cause a rapid increase in the stability of ion–uranyl carbonate complexes. Although Sample 3 had 30 times more calcium than Sample 2, the solutions were also undersaturated with respect to gypsum and calcite (SI = -1) until the end of the experiment (in the calculations, pH 6.8 and Eh -200 mV). We can assume that calcium in solid precipitates is predominantly in the form of whitlockite and hydroxylapatite phosphates. The point in
Figure 7C is very interesting (pH 6.8, Eh -100 mV). Computations showed that here, the solutions became saturated with pyrite and remaining sulphate. At pH > 6.8 and Eh -200 mV, the solutions were highly supersaturated towards pyrite, and gypsum dissolved (only sulphide was present in the solution). In the experiment, this reaction took place on the 180th day.
It is well known that biological processes depend on the redox of electron donor and acceptor pairs [
51,
52]. Obviously, with a shift in Eh from oxidizing conditions to anoxic conditions, the reduction of nitrate occurs first (as in our experiment), followed by Mn and then the Fe(OH)
3/Fe
2+ pair (reduction of Fe(III)). Sulphur reduction should occur after iron reduction, and uranium reduction is highly dependent on the presence of uranium carbonate complexes. In low-CO
2 media (black arrow), the UO
22+/U
4+ equilibrium lies in the zone of iron reduction, and there is a dramatic shift in the UO
2(CO
3)
34−/U
4+ equilibrium in the direction of decreasing Eh to a region close to that of methanogenesis.
Thus, it becomes clear why the reduced sulphide forms of Fe(II) generated by sulphate-reducing bacteria are extremely important for systems containing uranium. When oxidizing conditions change to reducing conditions during oxygen consumption and nitrate-sulphate reduction [
11], iron and manganese are first reduced to pyrite FeS
2, mackinawite, FeS
0.9, and alabandine MnS and then to uranyl ions [
53]. A reduction buffer was therefore formed to prevent possible reoxidation of uranium in the system.
The calculation considering a possible increase in pH, as a consequence of the denitrification process, revealed the main difference in the appearance of carbonates among the precipitates, primarily calcite. No significant changes were found in the uranium phases. It should be pointed out that in the natural conditions of contact between solutions and rocks, minerals will serve as a buffer for increased alkalinity.
In our laboratory experiments, the required database was obtained for the aquifer bioremediation process near NCCP. However, after the effective experimental tests of in situ uranium bioremediation processes at different objects, we assume that laboratory modelling of the biotreatment at the first stage of the work is a key to the successful completion of bioremediation. It should be added that the results obtained in our computations are consistent with those in a number of field experiments [
54,
55,
56,
57,
58,
59].
The formation of various biogenic mineral phases—phosphate uranium precipitates, calcite, pyrite FeS
2, and mackinawite—has been reported in many publications. The formation of biogenic ferrous deposits in the bioremediation of uranium pollution not only provides the immobilization conditions of uranium by the sorption mechanisms on new phases but also promotes their further mineralization and stronger fixation [
60,
61], including the inhibition of subsequent oxidation [
62]. According to numerous papers, microbial biofilms contribute to the mineralization of ferrous minerals, in particular haematite and pyrite [
63,
64,
65].


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}