
Probing the Interactions of LRP1 Ectodomain-Derived Peptides with Fibrillar Tau Protein and Its Impact on Cellular Internalization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Homology Modeling of LRP1
2.2.2. ClusPro
2.2.3. PrankWeb
2.2.4. Peptide Design
2.2.5. I-TASSER Studies
2.2.6. Molecular Docking
2.2.7. Molecular Dynamics Simulations
2.2.8. MMGBSA Energy Calculations
2.3. Laboratory Methods
2.3.1. E. coli Culture
2.3.2. DNA Purification and Extraction
2.3.3. DNA Quantification
2.3.4. Mammalian Cell Culture
2.3.5. Mammalian Cell Transfection
2.3.6. Preparation of Tau Fibrils
2.3.7. Thioflavin-T Assay
2.3.8. Antibody Labeling
2.3.9. Tau Internalization Assay
2.4. Characterization
2.4.1. Dynamic Light Scattering
2.4.2. Surface Plasmon Resonance
2.4.3. Circular Dichroism Spectroscopy
2.4.4. Fluorescence Spectroscopy
2.4.5. UV–Vis Spectroscopy
2.4.6. AFM
2.4.7. Flow Cytometry Analysis
3. Results and Discussion
3.1. Homology Modeling
3.2. Binding Interactions with PHF Tau
3.3. I-TASSER Studies
3.4. Molecular Docking Studies
3.4.1. Docking Studies with PHF Tau
3.4.2. Docking Studies with Additional Ligands Implicated in Alzheimer’s Disease
3.5. Molecular Dynamics Studies
MMBGSA Studies
3.6. Formation of Tau Fibrils
3.7. Binding Interactions of Fibrillar Tau with TauRP1-14 Peptide
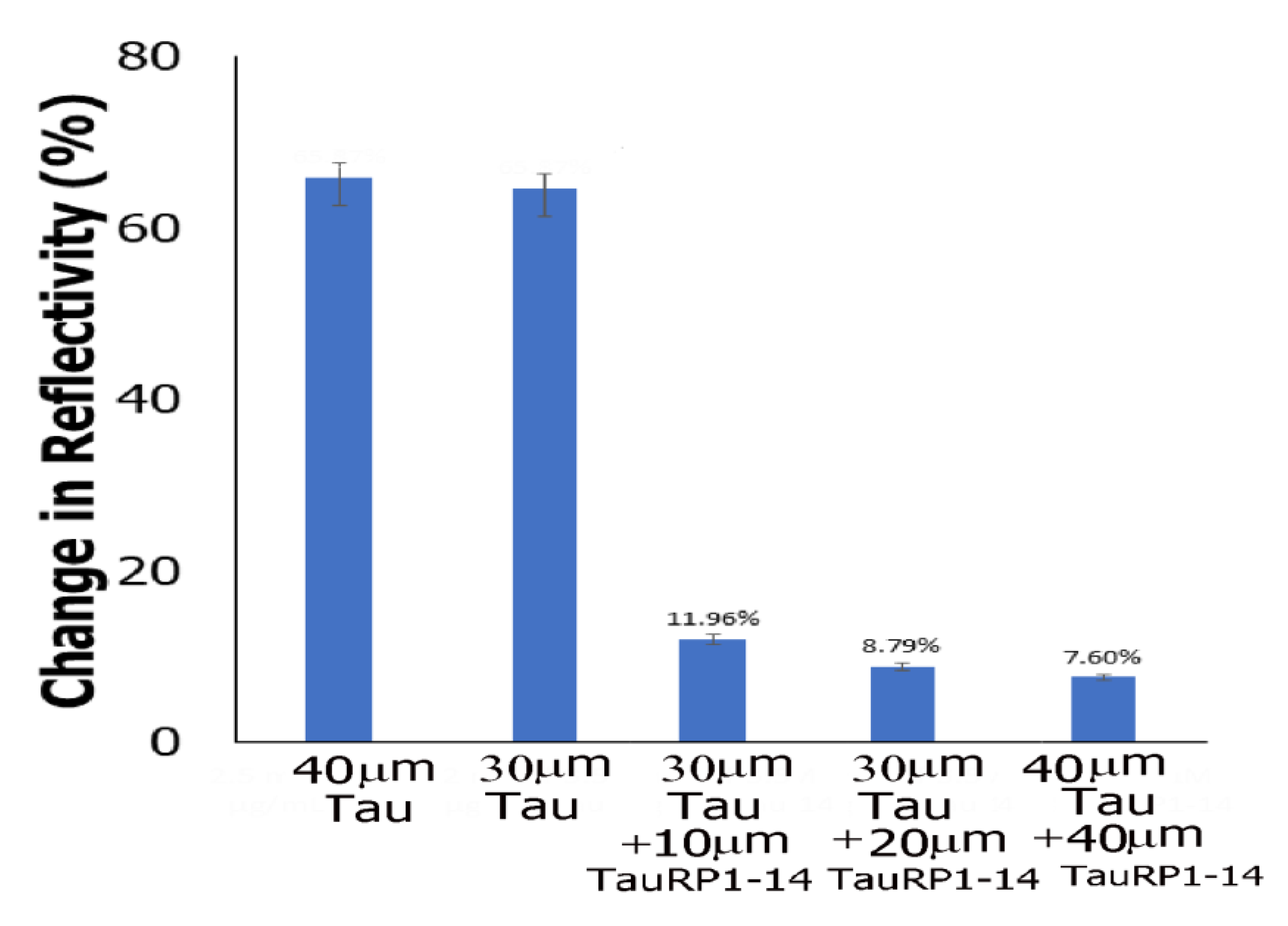
3.7.1. AFM
3.7.2. Circular Dichroism Studies
3.7.3. Surface Plasmon Resonance Studies
3.8. Flow Cytometry Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cleveland, D.; Hwo, S.; Kirschner, M. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Duan, A.R.; Jonasson, E.M.; Alberico, E.O.; Li, C.; Scripture, J.P.; Miller, R.A.; Alber, M.S.; Goodson, H.V. Interactions between Tau and different conformations of Tubulin: Implications of Tau function and mechanism. J. Mol. Biol. 2017, 429, 1424–1438. [Google Scholar] [CrossRef]
- Castro, T.G.; Muntenau, F.-D.; Cavaco-Pauolo, A. Electrostatics of Tau protein by molecular dynamics. Biomolecules 2019, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Castro, T.G.; Ferrerira, T.; Matama, T.; Munteanu, F.-D.; Cavaco-Paulo, A. Acetylation and phosphorylation processes modulate Tau’s binding to microtubules: A molecular dynamics study. Biochim. Biophys. Acta BBA-Gen. Subj. 2022, 1867, 130276. [Google Scholar] [CrossRef]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Briner, A.; Gotz, J.; Polanco, J. Fyn kinase controls Tau aggregation in vivo. Cell Rep. 2020, 32, 108045. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Guerrero-Munoz, M.; Castillo-Carranza, D.; Lasagna-Reeves, C.; Gerson, J.; Paulucci-Holthauzen, A.; Krishnamurthy, S.; Farhed, M.; Jackson, J.; Kayed, R. Pathological interface between oligomeric alpha-synuclein and tau in synucleinophathies. Biol. Psychiatry 2015, 78, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Mukrasch, M.; Bibow, J.; Blackledge, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of Tau to microtubules. Biochemistry 2009, 48, 10047–10055. [Google Scholar] [CrossRef]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Crowther, R. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. USA 1991, 88, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.; Phillips, C.; McCarty, J. Insight into seeded Tau fibril growth from molecular dynamics simulation of the Alzhiemer’s disease protofibril core. Front. Mol. Biosci. 2021, 8, 624302. [Google Scholar]
- Yamada, K. Extracellular Tau and its potential role in the propagation of tau pathology. Front. Neurosci. 2017, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.D.; Wassmer, T.; Graser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular monomeric and aggregated Tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, B.; DeVos, S.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.; Brodsky, F.; Marsa, J.; Bagchi, D.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Bobuhara, C.K.; et al. Neuronal uptake and propagation of rare phosphorylated high-molecular weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jurado, S. AMPA receptor trafficking in natural and pathological aging. Front. Mol. Neurosci. 2017, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- Morozova, V.; Cohen, L.; Makki, A.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A. Normal and pathological tau uptake mediated by M1/M3 muscarinic receptors promotes opposite neuronal changes. Front Cell Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Rubio, A.; Miras-Protugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 receptors in neuronal cells. Mol. Cell. Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef]
- Herbst, S.; Lewis, P.A.; Morris, H.R. The emerging role of LRRK2 in taupathies. Clin. Sci. 2022, 136, 1071–1079. [Google Scholar] [CrossRef]
- Evans, L.D.; Strano, A.; Campbell, A.; Karakoc, E.; Lorio, F.; Bassett, A.R.; Livesey, F.J. Whole genome CRISPR screens identify LRRK2-regulated endocytosis as a major mechanism for extracellular tau uptake by human neurons. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pensalfaini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.H.; Darji, S.; Pawlik, M.; et al. Endosomal dysfunction induced by directly overactivating Rab5 recapitulates prodromal and neurodegenerative features of Alzheimer’s disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.-C.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of β-amyloid induced tau pathology: The long and “folded” road to understand the mechanism. Mol. Neurodegen. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, A.; Chua, S.W.; Bertz, J.; Vokkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site specific phosphorylation of tau inhibits amyloid beta toxicity in Alzhiemer’s disease. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bok, E.; Leem, E.; Lee, B.-R.; Lee, J.; Yoo, C.; Lee, E.; Kim, J. Role of the lipid membrane and membrane proteins in tau pathology. Front. Cell Dev. Biol. 2021, 9, 653815. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M. Tau immunotherapies for Alzheimer’s disease and related Tauopathies: Progress and potential pitfalls. J. Alzheimer’s Dis. 2018, 66, 855–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilkova, M.; Nolle, A.; Kovacech, B.; Konteskova, E.; Weisova, P.; Filipcik, P.; Skarbana, R.; Prcina, M.; Hromadka, T.; Cehlar, O.; et al. Humanized tau antiboides promote tau uptake by human microglia without any increase of inflammation. Acta Neuropathol. Commun. 2020, 8, 74. [Google Scholar] [CrossRef]
- Rauch, J.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Bres, E.; Faissner, A. Low density receptor-related protein 1 interactions with the extracellular matrix: More than meets the eye. Front. Cell Dev. Biol. 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Lathuiliere, A.; Migliorini, M.; Arai, A.; Wani, M.; Dujardin, S.; Muraoglu, S.; Hyman, B.; Strickland, D. Regulation of tau internalization, degradation and seeding by LRP1 reveals multiple pathways of tau catabolism. J. Biol. Chem. 2021, 296, 100715. [Google Scholar] [CrossRef]
- Potere, N.; Del Buono, M.; Mauro, A.; Abbate, A.; Toldo, S. Low density lipoprotein receptor-related protein-1 in cardiac inflammation and infarct healing. Front. Cardiovasc. Med. 2019, 6, 51. [Google Scholar] [CrossRef]
- Neels, J.; van Den Berg, B.; Lookene, A.; Olivecrona, G.; Pannekoek, H.; van Zonneveld, A. The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J. Biol. Chem. 1999, 274, 31305–31311. [Google Scholar] [CrossRef] [PubMed]
- Fouët, G.; Gout, E.; Wicker-Planquart, C.; Bally, I.; de Nardis, C.; Dedieu, S.; Chouquet, A.; Gaboriaud, C.; Thielens, N.; Kleman, J.; et al. Complement C1q Interacts With LRP1 Clusters II and IV Through a Site Close but Different From the Binding Site of Its C1r and C1s-Associated Proteases. Front. Immunol. 2020, 11, 583754. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Lathuiliere, A.; Migliorini, M.; Arai, A.; Wani, M.; Dujardin, S.; Muratoglu, S.; Hyman, B.; Strickland, D. LRP1 and SORL1 regulate tau internalization and degradation and enhance tau seeding. bioRxiv 2020, 11, 386581. [Google Scholar]
- Herz, J.; Hamann, U.; Rogneo, S.; Myklebost, O.; Gausepohl, H.; Stanley, K. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. Embo. J. 1988, 7, 4119–4127. [Google Scholar] [CrossRef]
- Marakasova, E.; Olivares, P.; Karnaukhova, E.; Chun, H.; Hernandez, N.; Kurasawa, J.; Hassink, G.; Shestopal, S.; Strickland, D.; Sarafanov, A. Molecular chaperone RAP interacts with LRP1 in a dynamic bivalent mode and enhances folding of ligand-binding regions of other LDLR receptors. J. Biol. Chem. 2021, 297, 100842. [Google Scholar] [CrossRef]
- Fisher, C.; Beglova, N.; Blacklow, S. Structure of an LDLR-RAP Complex Reveals a General Mode for Ligand Recognition by Lipoprotein Receptors. Mol. Cell 2006, 22, 277–283. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Z.; Lu, S.; He, Q.; Deng, N.; Meng, H.; Pan, C.; Li, H.; Liu, M.; Huang, A.; et al. In-silico analysis of ligand-receptor binding patterns of alpha-MMC, TCS and MAP30 protein to LRP1 receptor. J. Mol. Graph. Model. 2020, 98, 107619. [Google Scholar] [CrossRef]
- Muhammed, M.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548. [Google Scholar] [CrossRef]
- Yu, Z.; Kang, L.; Zhao, W.; Wu, S.; Ding, L.; Zheng, F.; Liu, J.; Li, J. Identification of novel umami peptides from myosin via homology modeling and molecular docking. Food Chem. 2021, 344, 128728. [Google Scholar] [CrossRef]
- Nikolaev, D.; Shtyrov, A.; Panov, M.; Jamal, A.; Chakchir, O.; Kochemirovsky, V.; Olivucci, M.; Ryazantsev, M. A Comparative Study of Modern Homology Modeling Algorithms for Rhodopsin Structure Prediction. ACS Omega 2018, 3, 7555–7566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.; Falcon, B.; He, S.; Murzin, A.; Murshudov, G.; Garringer, H.; Crowther, R.; Ghetti, B.; Goedert, M.; Scheres, S. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Costales, P.; Fuentes-Prior, P.; Castellano, J.; Revuelta-Lopez, E.; Corral-Rodríguez, M.; Nasarre, L.; Badimon, L.; Llorente-Cortes, V. K Domain CR9 of Low Density Lipoprotein (LDL) Receptor-related Protein 1 (LRP1) Is Critical for Aggregated LDL-induced Foam Cell Formation from Human Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 14852–14865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmonsef, A. Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt. J. Med. Hum. Genet. 2019, 20, 6. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kelley, L.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Comeau, S.; Gatchell, D.; Vajda, S.; Camacho, C. ClusPro: A fully automated algorithm for protein–protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.; Xia, B.; Porter, K.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2020, 50, W593–W597. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homology proteins by coupling deep-learning contact maps with I-Tasser assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function predictions. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-Tasser Server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Eberhard, J.; Santos-Martins, D.; Tillack, A.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β (1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verderame, J.R.; Kantardjieff, K.; Segelke, B.; Weisgraber, K.; Rupp, B. Apolipoprotein E4, 22k Domain. Available online: https://www.rcsb.org/structure/1GS9 (accessed on 28 December 2022).
- Johnson, A.R.; Pavlovsky, A.G.; Ortwine, D.F.; Prior, F.; Man, C.F.; Bornemeier, D.A.; Bantoai, C.A.; Mueller, W.T.; McConnell, P.; Yan, C.; et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without fibroplasia side effects. J. Biol. Chem. 2007, 282, 27781–27791. [Google Scholar] [CrossRef] [Green Version]
- Durham, T.B.; Klimkowski, V.J.; Rito, C.J.; Marimuthu, J.; Toth, J.L.; Liu, C.; Durbin, J.D.; Stot, S.L.; Adams, L.; Swearingen, C.; et al. Identification of Potent and Selective Hydantoin Inhibitors of Aggrecanase-1 and Aggrecanase-2 That Are Efficacious in Both Chemical and Surgical Models of Osteoarthritis. Med. Chem. 2014, 57, 10476–10485. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL 2.1.1; Schrödinger, LLC.: New York, NY, USA, 2020. [Google Scholar]
- Bowers, K.; Chow, D.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Kleipeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.; Dahlgren, M.; Russell, E.; Bergen, C.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Jorgensen, W.; Maxwell, D.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Sastry, M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comp. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kobayashi, C.; Sugita, Y. Optimal temperature evaluation in molecular dynamics simulations with large time step. J. Chem. Theory Comput. 2019, 15, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.J.; Dueck, D. Clustering by passing messages between data points. Science 2007, 315, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onufriev, A.; Case, D. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Jacobson, M.; Friesner, R.; Xiang, Z.; Honig, B. On the Role of Crystal Packing Forces in Determining Protein Sidechain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.; Pincus, R.; Rapp, C.; Day, T.; Honing, B.; Shaw, D.; Friesner, R. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Clemente, R.; Igeño, M.I.; Población, A.G.; Guijo, M.I.; Merchán, F.; Blasco, R. Study of pH Changes in Media during Bacterial Growth of Several Environmental Strains. Proceedings 2018, 2, 1297. [Google Scholar]
- Giufrè, M.; Monaco, M.; Accogli, M.; Pantosti, A.; Cerquetti, M.; PAMURSA Study Group; Farina, C.; Fazii, P.; Mattei, R.; Moro, M.L.; et al. Emergence of the colistin resistance mcr-1determinant in commensal Escherichia coli from residents of long-term-care facilities in Italy. J. Antimicrob. Chemother. 2016, 71, 2329–2331. [Google Scholar] [CrossRef] [Green Version]
- Malinen, M.; Ali, I.; Bezençon, J.; Beaudoin, J.; Brouwer, K. Organic solute transporter OSTα/β is overexpressed in nonalcoholic steatohepatitis and modulated by drugs associated with liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G597–G609. [Google Scholar] [CrossRef]
- Rauch, J.; Chen, J.; Sorum, A.; Miller, G.; Sharf, T.; See, S.; Hsieh-Wilson, L.; Kampmann, M.; Kosik, K. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef] [Green Version]
- Christine, X.; Yuwen, L.; Dennis, C.; Zhefeng, G. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar]
- Biancalana, M.; Koide, S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Hunka, J.; Riley, J.; Debes, G. Approaches to overcome flow cytometry limitations in the analysis of cells from veterinary relevant species. BMC Vet. Res. 2020, 16, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, P.; Shen, Y.; Snavely, M.; Hiraga, K. A Cell-Based Internalization and Degradation Assay with an Activatable Fluorescence-Quencher Probe as a Tool for Functional Antibody Screening. J. Biomol. Screen. 2015, 20, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Minton, A. Recent applications of light scattering measurement in the biological and biopharmaceutical science. Anal. Biochem. 2016, 501, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Yanase, Y.; Hiragun, T.; Ishii, K.; Kawaguchi, T.; Yanase, T.; Kawai, M.; Sakamoto, K.; Michihiro, H. Surface Plasmon Resonance for Cell-Based Clinical Diagnosis. Sensors 2014, 14, 4948–4959. [Google Scholar] [CrossRef]
- Wang, S.; Boussaad, S.; Tao, N. Surface plasmon resonance enhanced optical absorption spectroscopy for studying molecular adsorbates. Rev. Sci. Instrum. 2001, 72, 3055. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Ollesch, J.; Wille, H.; Diamond, M. Conformational Diversity of Wild-type Tau Fibrils Specified by Templated Conformation Change. J. Biol. Chem. 2008, 284, 3546–3551. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Galliano, M.; Toulza, E.; Jonca, N.; Gonias, S.; Serre, G.; Guerrin, M. Binding of α2ML1 to the Low Density Lipoprotein Receptor-Related Protein 1 (LRP1) Reveals a New Role for LRP1 in the Human Epidermis. PLoS ONE 2008, 3, e2729. [Google Scholar] [CrossRef] [Green Version]
- Lillis, A.; van Duyn, L.; Murphy-Ullrich, J.; Strickland, D. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudenko, G.; Henry, L.; Henderson, K.; Ichtchenko, K.; Brown, M.; Goldstein, J.; Deisenhofer, J. Structure of the LDL receptor extracellular domain at endosomal pH. Science 2002, 298, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Ittisoponpisan, S.; Islam, S.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Williams, B. Low-Density Lipoprotein Receptor-Related Proteins in Skeletal Development and Disease. Physiol. Rev. 2017, 97, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Blacklow, S. Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Paredes Lopez, O.; Parhiz, H.; et al. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Hamley, I.W. Small bioactive peptides for biomaterials design and therapeutics. Chem. Rev. 2017, 117, 14015–14041. [Google Scholar] [CrossRef] [Green Version]
- Perlikowska, R. Whether short peptides are good candidates for future neuroprotective therapeutics? Peptides 2021, 140, 170528. [Google Scholar] [CrossRef]
- Naranda, T.; Wong, K.; Kaufman, R.; Goldstein, A.; Olsson, L. Activation of erythropoietin receptor in the absence of hormone by a peptide that binds to a domain different from the hormone binding site. Proc. Natl. Acad. Sci. USA 2021, 96, 7569–7574. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.; Alhossary, A.; Mu, Y.; Kwoh, C. Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [Green Version]
- Nacharaju, P.; Ko, L.; Yen, S. Characterization of in vitro glycation sites of tau. J. Neurochem. 1997, 69, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Jiménez, J.; Sayas, C.; Bolós, M.; Zabala, J.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonias, S.L.; Campana, W.M. LDL Receptor-related protein 1. Am. J. Pathol. 2014, 184, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Kimura, K.; Haneda, M.; Ishida, Y.; Sawada, M.; Isobe, K.-I. Induction of matrix metalloproteinases (MMP3, MMP12 and MMP13) expression in the microglia by amyloid-β stimulation via the PI3K/Akt pathway. Exp. Gerontol. 2007, 42, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Guress, M.S.; Ural, M.N.; Gulec, M.A.; Akyol, O.; Akyol, S. Pathophysiological function of ADAMTS enzymes on molecular mechanisms of Alzheimer’s disease. Aging Dis. 2016, 7, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Hashimoto, T.; Yabuki, C.; Nagae, Y.; Tachikawa, M.; Strickland, D.K.; Liu, Q.; Bu, G.; Basak, J.M.; Holtzman, D.M.; et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J. Biol. Chem. 2008, 283, 34554–34562. [Google Scholar] [CrossRef] [Green Version]
- Walter, S.; Jumpertz, T.; Hutternrauch, M.; Ogoerk, I.; Gerber, H.; Storck, S.E.; Zampar, S.; Dimitrov, M.; Lehmann, S.; Lepka, K.; et al. The metalloprotease ADAMTS4 generates N-truncated Aβ-x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [Green Version]
- Calandra, S.; Tarugi, P.; Speedy, H.; Dean, A.; Bertolini, S.; Shoulders, C. Mechanisms and genetic determinants regulating sterol absorption, circulating LDL levels and sterol elimination: Implications for classification and disease risk. J. Lipid Res. 2011, 52, 1885–1926. [Google Scholar] [CrossRef] [Green Version]
- Ahyayauch, H.; Raab, M.; Busto, J.V.; Andraka, N.; Arrondo, J.-L.R.; Masserini, M.; Tvaroska, I.; Goni, F.M. Binding of β-amyloid (1-42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: Modeling and experimental studies. Biophys. J. 2012, 103, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, D.J.; Wesén, E.; Bjorkeroth, J.; Rocha, S.; Esbjorer, E.K. Lipid membranes catalyse the fibril formation of the amyloid-β (1-42) peptide through lipid-fibril interactions that reinforce secondary pathways. Biochim. Biophys. Acta. Biomembranes 2017, 1859, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, B.; Strock, S.E.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.S.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7234–7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Do, T.; Larini, L.; LaPointe, N.; Sercel, A.; Shade, M.; Feinstein, S.; Bowers, M.; Shea, J.-E. Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J. Phys. Chem. B. 2015, 119, 4582–4593. [Google Scholar] [CrossRef] [Green Version]
- Lobanov, M.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration is indicator of compactness of protein structure. Mol. Biol. 2008, 42, 701–706. [Google Scholar] [CrossRef]
- Durham, E.; Dorr, B.; Woetzel, N.; Staritzbichler, R.; Meiler, J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J. Mol. Model. 2009, 15, 1093–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods: II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippens, G.; Sillen, A.; Landrieu, I.; Amniai, L.; Sibille, N.; Barbier, P.; Leroy, A.; Hanoulle, X.; Wieruszeski, J. Tau aggregation in Alzheimer’s disease: What role for phosphorylation? Prion 2007, 1, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Jung, Y.J.; Chinnathambi, S.; Madelkow, E.-M.; Mandelkow, E.; Muller, D. Human Tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J. Biol. Chem. 2010, 285, 27302–27313. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular Tau Oligomers as Intermediates of Tau Filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Morozova, O.A.; March, Z.M.; Robinson, A.S.; Colby, D.W. Conformational features of tau fibrils from Alzhiemer’s disease brain are faithfully propagated recombinant protein. Biochemistry 2013, 52, 6960–6967. [Google Scholar] [CrossRef] [Green Version]
- Chemes, L.B.; Alonso, L.G.; Noval, M.G.; de Prat-Gay, G. Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol. Biol. 2012, 895, 387–404. [Google Scholar] [PubMed]
- Sakamoto, K.; Shinohara, T.; Adachi, Y.; Asami, T.; Ohtaki, T. A novel LRP1-binding peptide L57 that crosses the blood brain barrier. Biochem. Biophys. Rep. 2017, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Migliorini, M.; Arai, A.; Catania, S.; Hyman, B.; Strickland, D. LRP1 mediates tau endocytosis in a process that is modulated by apolipoprotein E: Molecular and cell biology/receptors. Alzheimer’s Dement. 2020, 16, e045959. [Google Scholar] [CrossRef]
- Demeule, M.; Regina, A.; Poirier, J.; Béliveau, R.; Ché, C.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P. Identification and Design of Peptides as a New Drug Delivery System for the Brain. J. Pharmacol. Exp. Ther. 2007, 324, 1064–1072. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.-C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pockets | Rank | Score | Probability |
|---|---|---|---|
| Pocket 1 | 1 | 83.27 | 0.993 |
| Pocket 2 | 2 | 82.55 | 0.993 |
| Pocket 3 | 3 | 28.54 | 0.908 |
| Sequence Abbreviation | Sequence Designed |
|---|---|
| TauRP1-1 | SKAWVCDGDN |
| TauRP1-2 | KAWVCDGDND |
| TauRP1-3 | AWVCDGDNDC |
| TauRP1-4 | WVCDGDNDCE |
| TauRP1-5 | VCDGDNDCED |
| TauRP1-6 | CDGDNDCEDN |
| TauRP1-7 | DGDNDCEDNS |
| TauRP1-8 | GDNDCEDNSD |
| TauRP1-9 | DNDCEDNSDE |
| TauRP1-10 | NDCEDNSDEE |
| TauRP1-11 | DCEDNSDEEN |
| TauRP1-12 | CEDNSDEENC |
| TauRP1-13 | EDNSDEENCE |
| TauRP1-14 | DNSDEENCES |
| Peptide | Secondary Structure | C-Score | Estimated TM-Score |
|---|---|---|---|
| 23-AA sequence from Cluster 2 Domain SKAWVCDGDNDCEDN-SDEENCES | CCCSSSCCCCCCCCCCCHHHCCC | 0.35 | 0.76 ± 0.10 |
| TauRP1-1 | CCCSSCCCCC | −0.59 | 0.64 ± 0.13 |
| TauRP1-2 | CCSSCCCCCC | −0.33 | 0.67 ± 0.13 |
| TauRP1-3 | CCCCCCCCCC | −0.15 | 0.69 ± 0.12 |
| TauRP1-4 | CCCCCCCCCC | −0.16 | 0.69 ± 0.12 |
| TauRP1-5 | CCCCCCCCCC | −0.76 | 0.62 ± 0.14 |
| TauRP1-6 | CCCCCCCCCC | −0.05 | 0.71 ± 0.12 |
| TauRP1-7 | CCCCCCCCCC | −0.30 | 0.67 ± 0.12 |
| TauRP1-8 | CCCCCCCCCC | 0.05 | 0.72 ± 0.11 |
| TauRP1-9 | CCCCCCCCCC | −0.60 | 0.64 ± 0.13 |
| TauRP1-10 | CCCCCCCCCC | 0.48 | 0.78 ± 0.10 |
| TauRP1-11 | CCCCCCCCCC | 0.07 | 0.72 ± 0.11 |
| TauRP1-12 | CCCCCCCCCC | −0.79 | 0.61 + 0.14 |
| TauRP1-13 | CCCCCCCCCC | −0.38 | 0.66 ± 0.13 |
| TauRP1-14 | CCCCHHCCCC | −0.30 | 0.67 ± 0.12 |
| Sequence Abbreviation | Mode 1 | Mode 2 | Mode 3 | Mode 4 | Mode 5 | Mode 6 | Mode 7 | Mode 8 | Mode 9 |
|---|---|---|---|---|---|---|---|---|---|
| TauRP1-2 |  −4.6 | −4.5 | −4.5 | −4.4 | −4.4 |  −4.3 | −4.2 | −4.2 | −4.1 |
| TauRP1-4 | −4.4 | −4.4 | −4.3 | −4.3 | −4.3 | −4.2 | −4.2 | −4.1 | −4.1 |
| TauRP1-6 | −4.9 | −4.9 | −4.8 | −4.8 | −4.8 | −4.8 | −4.7 | −4.7 | −4.7 |
| TauRP1-7 | −4.4 | −4.2 | −4.2 | −4.1 | −4.0 | −4.0 | −3.9 | −3.9 | −3.9 |
| TauRP1-8 | −5.3 | −5.3 | −5.2 | −5.2 | −5.1 | −5.0 | −5.0 | −4.9 | −4.8 |
| TauRP1-13 | −4.7 | −4.5 | −4.5 | −4.4 | −4.3 | −4.3 | −4.2 | −4.2 | −4.2 |
| TauRP1-14 | −5.8 | −5.7 | −5.7 | −5.6 | −5.5 | −5.4 | −5.3 | −5.3 | −5.3 |
indicates that peptide was found outside the LRP1 binding pocket. indicates modes where the peptide bound within the predicted binding pocket.| Name of Peptide | Average ΔG Binding Energy (kcal/mol) | Average Electrostatic Energy (kcal/mol) | Average H-Bond Energy (kcal/mol) | Average Lipophilic Energy (kcal/mol) | Average vdW Energy (kcal/mol) |
|---|---|---|---|---|---|
| TauRP1-2 | −75.71 | 6.73 | −5.20 | −9.59 | −58.42 |
| TauRP1-4 | −53.43 | −215.64 | −5.01 | −8.74 | −43.89 |
| TauRP1-8 | −42.05 | −235.13 | −5.57 | −4.01 | −52.34 |
| TauRP1-14 | −70.70 | −342.01 | −10.17 | −5.61 | −53.01 |
| Secondary Structure | Tau Fibrils without TauRP1-14 | Tau Fibrils Pre-Associated with 10 µM TauRP1-14 | Tau Fibrils Pre-Associated with 20 µM TauRP1-14 | Tau Fibrils Pre-Associated with 40 µM TauRP1-14 |
|---|---|---|---|---|
| Helix | 23.1% | 1.50% | 0.00% | 17.8% |
| Antiparallel β-sheet | 17.8% | 31.5% | 37.5% | 14.6% |
| Parallel β-sheet | 26.5% | 0.00% | 0.00% | 0.00% |
| Turn | 24.7% | 13.9% | 14.1% | 12.0% |
| Disordered | 7.9% | 53.1% | 48.3% | 55.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boder, E.J.; Goncalves, B.G.; Lebedenko, C.G.; Banerjee, I.A. Probing the Interactions of LRP1 Ectodomain-Derived Peptides with Fibrillar Tau Protein and Its Impact on Cellular Internalization. Appl. Sci. 2023, 13, 853. https://doi.org/10.3390/app13020853
Boder EJ, Goncalves BG, Lebedenko CG, Banerjee IA. Probing the Interactions of LRP1 Ectodomain-Derived Peptides with Fibrillar Tau Protein and Its Impact on Cellular Internalization. Applied Sciences. 2023; 13(2):853. https://doi.org/10.3390/app13020853
Chicago/Turabian StyleBoder, E. Josephine, Beatriz G. Goncalves, Charlotta G. Lebedenko, and Ipsita A. Banerjee. 2023. "Probing the Interactions of LRP1 Ectodomain-Derived Peptides with Fibrillar Tau Protein and Its Impact on Cellular Internalization" Applied Sciences 13, no. 2: 853. https://doi.org/10.3390/app13020853
APA StyleBoder, E. J., Goncalves, B. G., Lebedenko, C. G., & Banerjee, I. A. (2023). Probing the Interactions of LRP1 Ectodomain-Derived Peptides with Fibrillar Tau Protein and Its Impact on Cellular Internalization. Applied Sciences, 13(2), 853. https://doi.org/10.3390/app13020853