Impact of Alloying on Stacking Fault Energies in γ-TiAl




Abstract
:1. Introduction
2. Methods
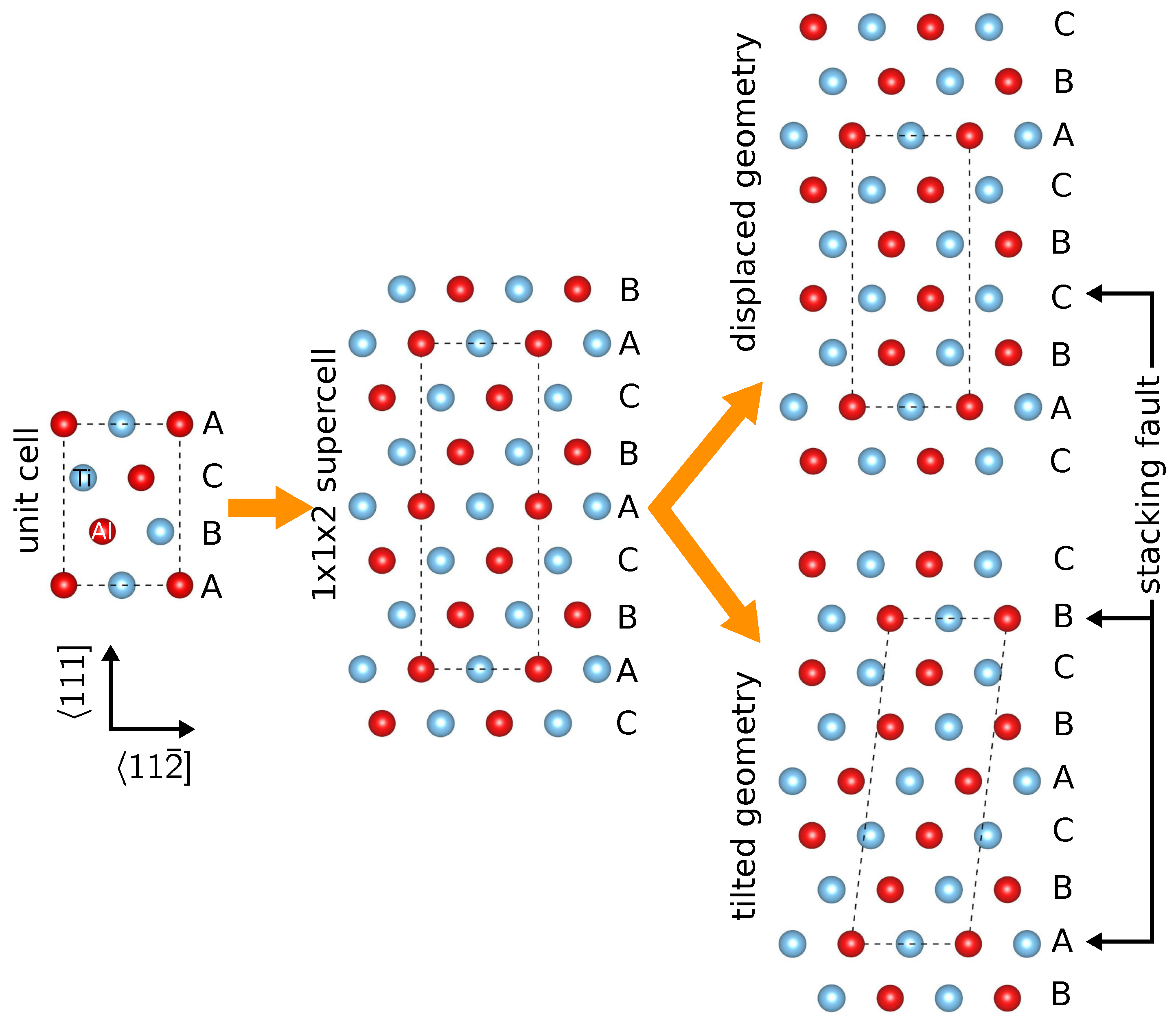
2.1. Geometry of Planar Defects in -TiAl
2.2. Modeling of SFs
2.3. Computational Details
3. Results and Discussion
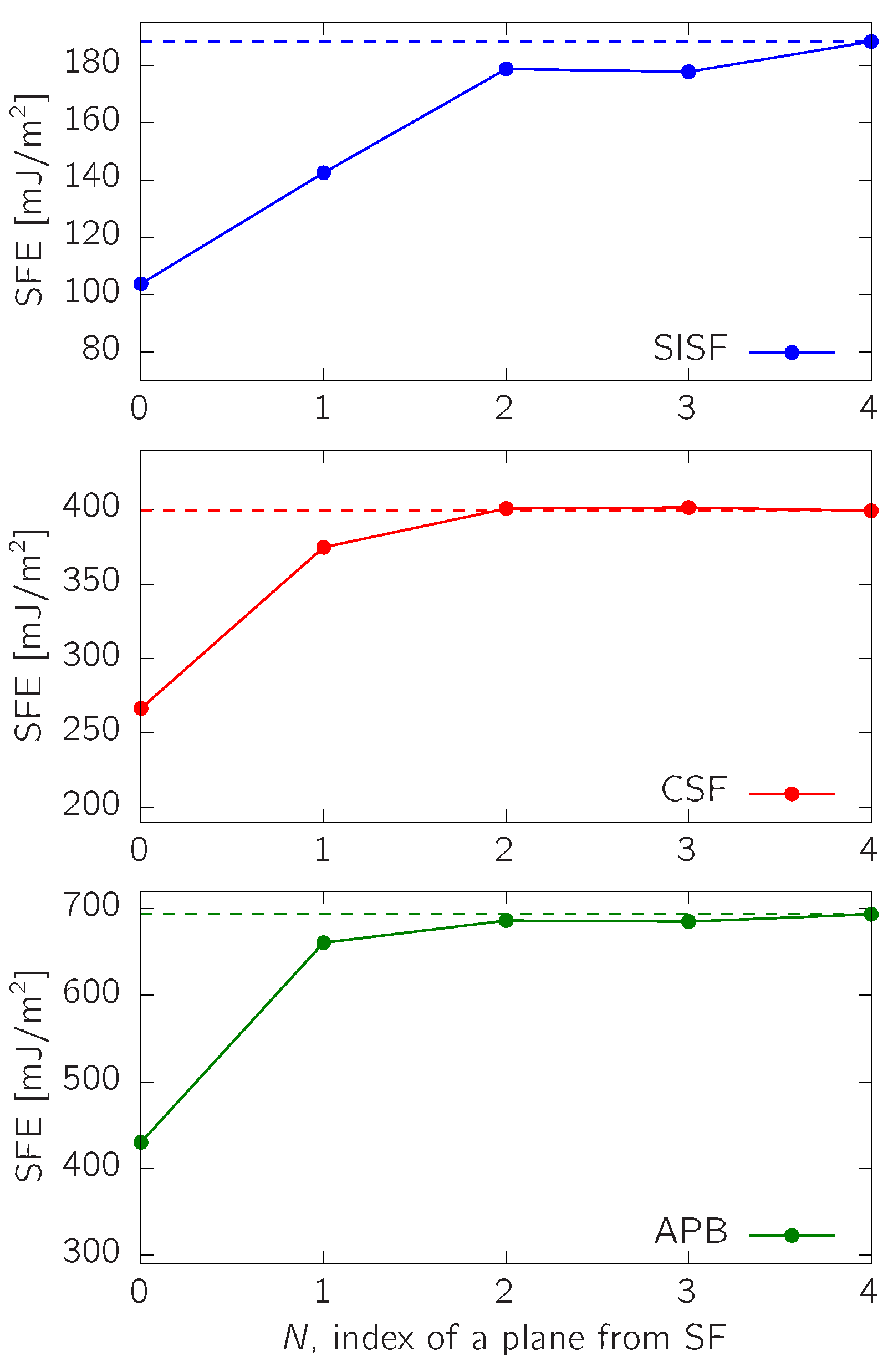
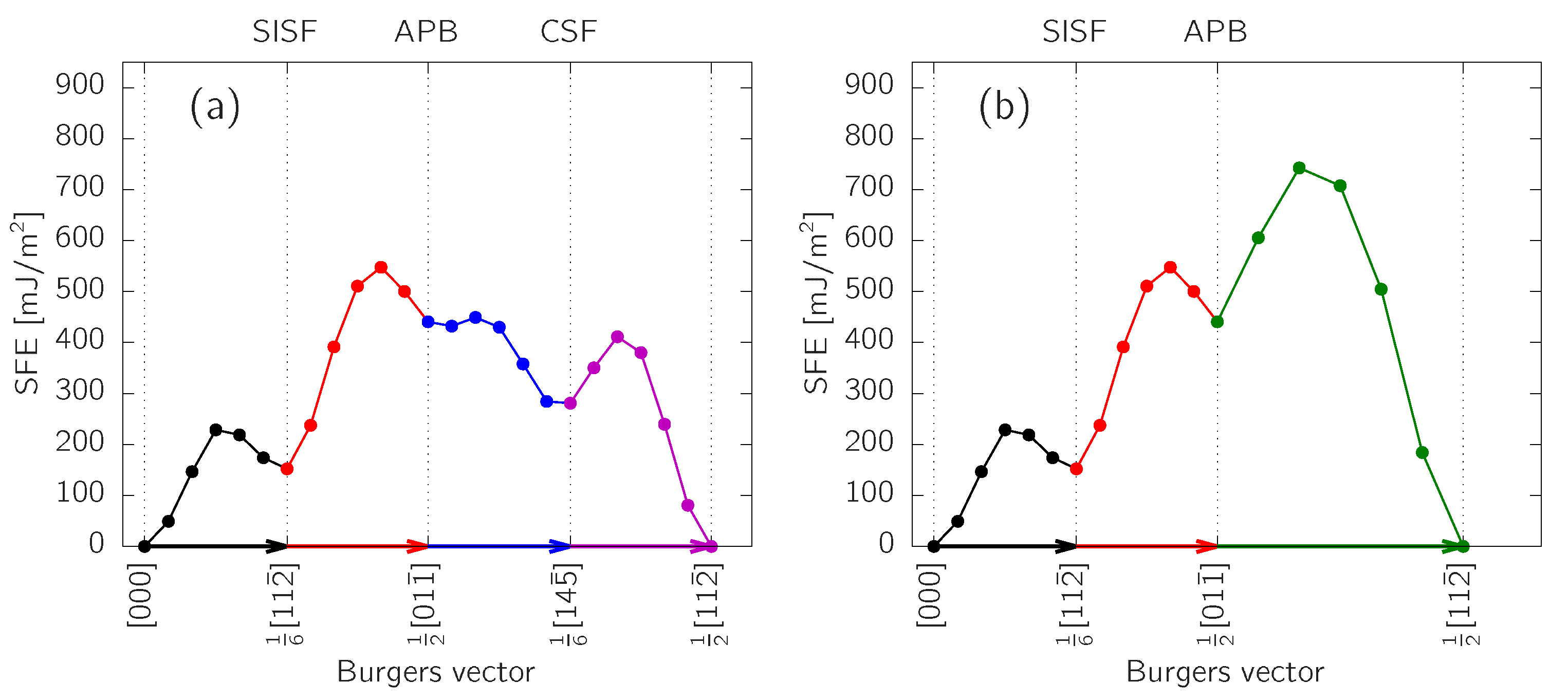
3.1. SFE in -TiAl
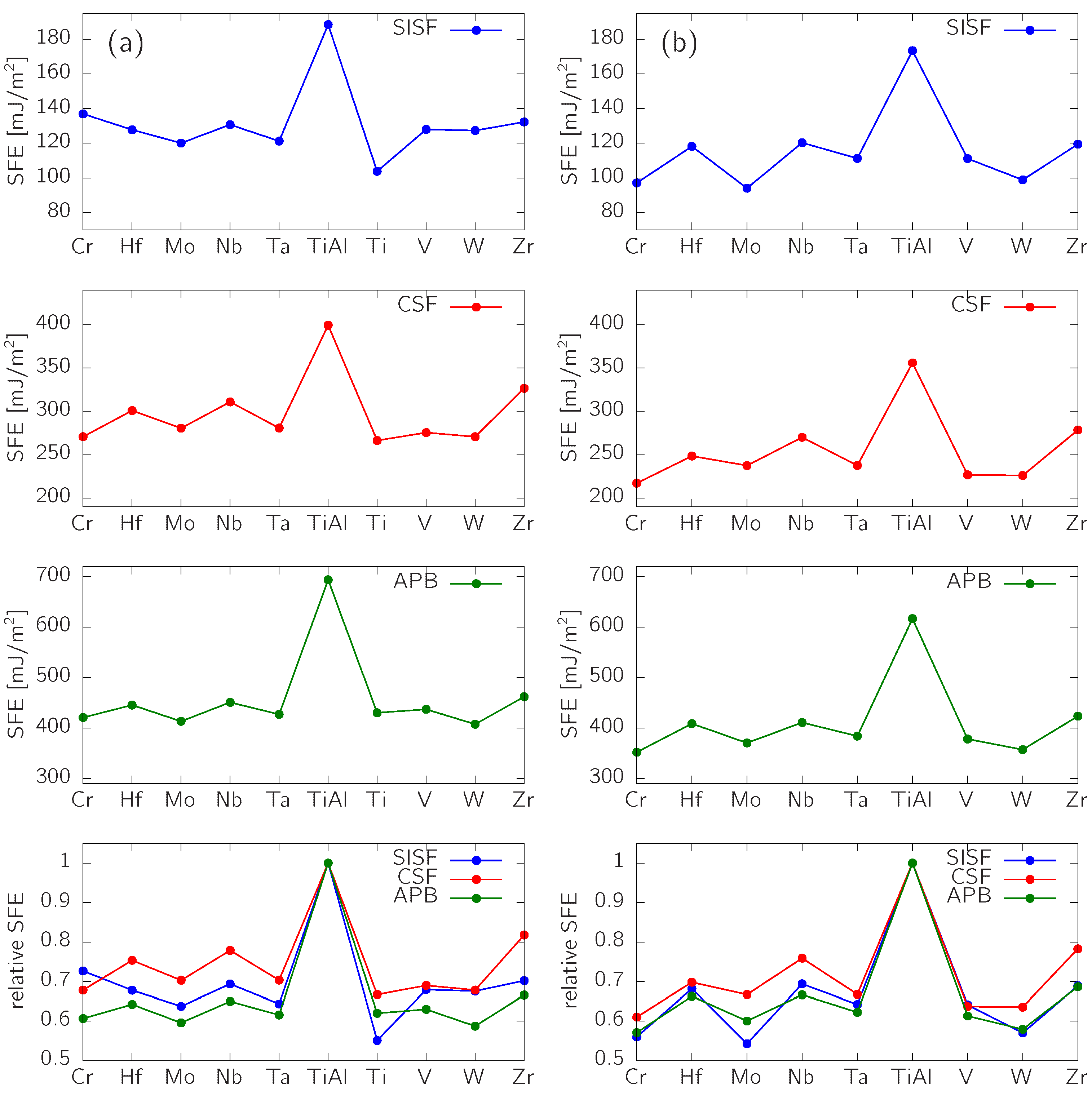
3.2. Impact of Alloying Elements
3.3. Comparison with Experiment
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| APB | anti-phase boundary |
| CASTEP | Cambridge Serial Total Energy Package |
| CPA | coherent potential approximation |
| CSF | complex stacking fault |
| DFT | density functional theory |
| ESF | extrinsic stacking fault |
| FP-LAPW | full-potential linearized augmented plane-wave (method) |
| FP-LMTO | full-potential linear muffin-tin orbital (method) |
| GGA | generalized gradient approximation |
| GSFE | generalized stacking fault energy |
| LDA | local density approximation |
| ISF | intrinsic stacking fault |
| SF | stacking fault |
| SFE | stacking fault energy |
| SISF | superlattice intrinsic stacking fault |
| TEM | transmission electron microscopy |
| TM | transition metal |
| VASP | Vienna Ab initio Simulation Package |
| xc | exchange and correlation (potential, effects) |
References
- Appel, F.; Paul, J.; Oehring, M. Gamma Titanium Aluminide Alloys: Science and Technology; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Clemens, H.; Mayer, S. Design, Processing, Microstructure, Properties, and Applications of Advanced Intermetallic TiAl Alloys. Adv. Eng. Mater. 2013, 15, 191–215. [Google Scholar] [CrossRef]
- Mayer, S.; Erdely, P.; Fischer, F.D.; Holec, D.; Kastenhuber, M.; Klein, T.; Clemens, H. Intermetallic β-Solidifying γ-TiAl Based Alloys—From Fundamental Research to Application. Adv. Eng. Mater. 2017, 19, 1600735. [Google Scholar] [CrossRef]
- Dimiduk, D.M. Gamma titanium aluminide alloys—An assessment within the competition of aerospace structural materials. Mater. Sci. Eng. A 1999, 263, 281–288. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Bernstein, N. A first-principles measure for the twinnability of FCC metals. J. Mech. Phys. Solids 2004, 52, 2507–2519. [Google Scholar] [CrossRef]
- Wiezorek, J.M.K.; Humphreys, C.J. On the hierarchy of planar fault energies in TiAl. Scr. Metall. Mater. 1995, 33, 451–458. [Google Scholar] [CrossRef]
- Yoo, M.H.; Fu, C.L. Physical constants, deformation twinning, and microcracking of titanium aluminides. Metall. Mater. Trans. A 1998, 29, 49–63. [Google Scholar] [CrossRef]
- Zhang, W.J.; Appel, F. Effect of Al content and Nb addition on the strength and fault energy of TiAl alloys. Mater. Sci. Eng. A 2002, 329–331, 649–652. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, H.W.; Zhao, X.N.; Meng, X.K.; Liu, Z.G.; Boll, T.; Al-Kassab, T. Dissociation of super-dislocations and the stacking fault energy in TiAl based alloys with Nb-doping. Phys. Lett. A 2006, 358, 231–235. [Google Scholar] [CrossRef]
- Vitek, V.; Ito, K.; Siegl, R.; Znam, S. Structure of interfaces in the lamellar TiAl: Effects of directional bonding and segregation. Mater. Sci. Eng. A 1997, 239, 752–760. [Google Scholar] [CrossRef]
- Ehmann, J.; Fähnle, M. Generalized stacking-fault energies for TiAl: Mechanical instability of the (111) antiphase boundary. Philos. Mag. A 1998, 77, 701–714. [Google Scholar] [CrossRef]
- Liu, Y.L.; Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-principles study of shear deformation in TiAl and Ti3Al. Intermetallics 2007, 15, 428–435. [Google Scholar] [CrossRef]
- Wen, Y.F.; Sun, J. Generalized planar fault energies and mechanical twinning in gamma TiAl alloys. Scr. Mater. 2013, 68, 759–762. [Google Scholar] [CrossRef]
- Kanani, M.; Hartmaier, A.; Janisch, R. Interface properties in lamellar TiAl microstructures from density functional theory. Intermetallics 2014, 54, 154–163. [Google Scholar] [CrossRef]
- Simmons, J.P.; Rao, S.I.; Dimiduk, D.M. Atomistics simulations of structures and properties of 1/2 dislocations using three different embedded-atom method potentials fit to γ-TiAl. Philos. Mag. A 1997, 75, 1299–1328. [Google Scholar] [CrossRef]
- Mahapatra, R.; Girshick, A.; Pope, D.P.; Vitek, V. Deformation mechanisms of near-stoichiometric single phase TiAl single crystals: A combined experimental and atomistic modeling study. Scr. Metall. Mater. 1995, 33, 1921–1927. [Google Scholar] [CrossRef]
- Kanani, M.; Hartmaier, A.; Janisch, R. Stacking fault based analysis of shear mechanisms at interfaces in lamellar TiAl alloys. Acta Mater. 2016, 106, 208–218. [Google Scholar] [CrossRef]
- Woodward, C.; Maclaren, J.M. Planar fault energies and sessile dislocation configurations in substitutionally disordered Ti-Al with Nb and Cr ternary additions. Philos. Mag. A 1996, 74, 337–357. [Google Scholar] [CrossRef]
- Vitek, V. Theory of the Core Structures of Dislocations in Body-Centered-Cubic Metals. Cryst. Lattice Defects 1974, 5, 1–34. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B Condens. Matter 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Clemens, H.; Mayer, S. Intermetallic titanium aluminides in aerospace applications—Processing, microstructure and properties. Mater. High Temp. 2016, 33, 560–570. [Google Scholar] [CrossRef]
- Jiang, C. First-principles study of site occupancy of dilute 3d, 4d and 5d transition metal solutes in L10 TiAl. Acta Mater. 2008, 56, 6224–6231. [Google Scholar] [CrossRef]
- Holec, D.; Reddy, R.K.; Klein, T.; Clemens, H. Preferential site occupancy of alloying elements in TiAl-based phases. J. Appl. Phys. 2016, 119, 205104. [Google Scholar] [CrossRef]
- Dumitraschkewitz, P. Planar Faults in γ-TiAl: An Atomistic Study. Master’s Thesis, Montanuniversität Leoben, Leoben, Austria, 2015. [Google Scholar]
- Zope, R.R.; Mishin, Y. Interatomic potentials for atomistic simulations of the Ti-Al system. Phys. Rev. B Condens. Matter 2003, 68, 024102. [Google Scholar] [CrossRef]
- Farkas, D.; Jones, C. Interatomic potentials for ternary Nb-Ti-Al alloys. Modell. Simul. Mater. Sci. Eng. 1999, 4, 23. [Google Scholar] [CrossRef]
- Thornton, P.R.; Mitchell, T.E.; Hirsch, P.B. The dependence of cross-slip on stacking-fault energy in face-centered cubic metals and alloys. Philos. Mag. 1962, 7, 1349–1369. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APB | CSF | SISF | Note | |
|---|---|---|---|---|
| present work | 717 | 415 | 188 | GGA-PW91, VASP, tilted supercells |
| 635 | 370 | 173 | GGA-PW91, VASP, tilted supercells, fully relaxed | |
| 711 | 414 | 188 | GGA-PW91, VASP, displaced supercells | |
| 694 | 392 | 179 | LDA, VASP, tilted supercells | |
| [10] | 710 | 314 | 134 | LDA, FP-LMTO, tilted supercells(?) |
| [11] | 756 | 420 | 184 | LDA, FP-LAPW, tilted supercells |
| [12] | 499 | 329 | 137 | GGA-PW91, CASTEP, tilted supercells |
| [13] | 355 | 184 | GGA-PW91, VASP, displaced supercells | |
| [14] | 663 | 400 | 170 | GGA-PW91, VASP, displaced supercells |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumitraschkewitz, P.; Clemens, H.; Mayer, S.; Holec, D. Impact of Alloying on Stacking Fault Energies in γ-TiAl. Appl. Sci. 2017, 7, 1193. https://doi.org/10.3390/app7111193
Dumitraschkewitz P, Clemens H, Mayer S, Holec D. Impact of Alloying on Stacking Fault Energies in γ-TiAl. Applied Sciences. 2017; 7(11):1193. https://doi.org/10.3390/app7111193
Chicago/Turabian StyleDumitraschkewitz, Phillip, Helmut Clemens, Svea Mayer, and David Holec. 2017. "Impact of Alloying on Stacking Fault Energies in γ-TiAl" Applied Sciences 7, no. 11: 1193. https://doi.org/10.3390/app7111193
APA StyleDumitraschkewitz, P., Clemens, H., Mayer, S., & Holec, D. (2017). Impact of Alloying on Stacking Fault Energies in γ-TiAl. Applied Sciences, 7(11), 1193. https://doi.org/10.3390/app7111193