Molecular Controlling the Transport Properties for Benzothiadiazole-Based Hole Transport Materials


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
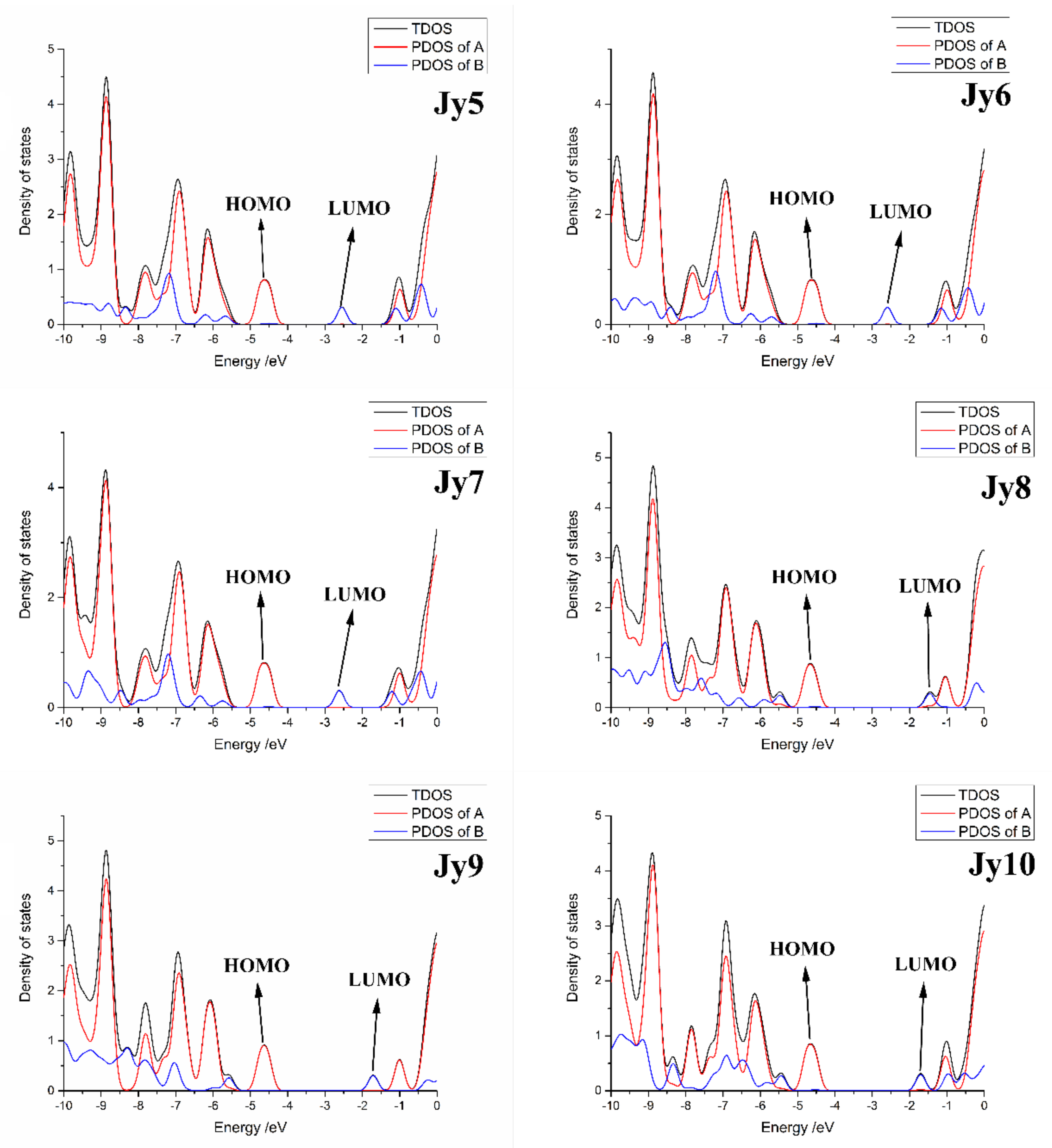
3.1. Ground-State Geometries and Frontier Molecular Orbitals
3.2. Ionization Potentials, Electron Affinities and Absolute Harness
3.3. Reorganization Energy of HTM Molecules
3.4. Optical Absorption Properties
3.5. Hole Mobility Rate and Hole Mobility of HTMs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, G.; Chang, W.H.; Yang, Y. Low-bandgap conjugated polymers enabling solution-processable tandem solar cells. Nat. Rev. Mater. 2017, 2, 17043. [Google Scholar] [CrossRef]
- Cheng, P.; Zhan, X.W. Stability of organic solar cells: Challenges and strategies. Chem. Soc. Rev. 2016, 45, 2544–2582. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Li, G.; Zhang, X.W.; Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics 2018, 12, 131–142. [Google Scholar] [CrossRef]
- Shan, C.; Gaoquan, S. Two-Dimensional Materials for Halide Perovskite-Based Optoelectronic Devices. Adv. Mater. 2017, 29, 1605448. [Google Scholar]
- Wolf, S.D.; Holovsky, J.; Moon, S.J.; Löper, P.; Niesen, B.; Ledinsky, M.; Haug, F.J.; Yum, J.H.; Ballif, C. Organometallic Halide Perovskites: Sharp Optical Absorption Edge and Its Relation to Photovoltaic Performance. J. Phys. Chem. Lett. 2014, 5, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.W.; Yu, Y.; Wang, C.L.; Liao, W.Q.; Shrestha, N.; Grice, C.R.; Cimaroli, A.J.; Guan, L.; Ellingson, R.J.; Zhu, K.; et al. Low-bandgap mixed tin–lead iodide perovskite absorbers with long carrier lifetimes for all-perovskite tandem solar cells. Nat. Energy 2017, 2, 17018. [Google Scholar] [CrossRef]
- Yang, W.S.; Park, B.W.; Jung, E.H.; Jeon, N.J.; Kim, Y.C.; Lee, D.U.; Shin, S.S.; Seo, J.; Kim, E.K.; Noh, J.H.; et al. Iodide management in formamidinium-lead-halide–based perovskite layers for efficient solar cells. Science 2017, 356, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.I.H.; Qurashi, A.; Nazeeruddin, M.K. Frontiers, opportunities, and challenges in perovskite solar cells: A critical review. J. Photochem. Photobiol. C Photochem. Rev. 2018, 35, 1–24. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Leijtens, T.; Eperon, G.E.; Stranks, S.D.; Nicholas, R.J.; Snaith, H.J. Carbon Nanotube/Polymer Composites as a Highly Stable Hole Collection Layer in Perovskite Solar Cells. Nano Lett. 2014, 14, 5561–5568. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.J.; Li, Q.S.; Li, Z.S. Exploring the electrochemical properties of hole transport materials with spiro-cores for efficient perovskite solar cells from first-principles. Nanoscale 2016, 8, 6146–6154. [Google Scholar] [CrossRef] [PubMed]
- Calil, L.; Kazim, S.; Gratzel, M.; Ahmad, S. Hole-Transport Materials for Perovskite Solar Cells. Angew. Chem. Int. Ed. 2016, 55, 14522–14545. [Google Scholar] [CrossRef] [PubMed]
- Hawash, Z.; Ono, L.K.; Qi, Y.B. Recent Advances in Spiro-MeOTAD Hole Transport Material and Its Applications in Organic–Inorganic Halide Perovskite Solar Cells. Adv. Mater. Interfaces 2018, 5, 1700623. [Google Scholar] [CrossRef]
- Xu, B.; Sheibani, E.; Liu, P.; Zhang, J.B.; Tian, H.N.; Vlachopoulos, N.; Boschloo, G.; Kloo, L.; Hagfeldt, A.; Sun, L.C. Carbazole-Based Hole-Transport Materials for Efficient Solid-State Dye-Sensitized Solar Cells and Perovskite Solar Cells. Adv. Mater. 2014, 26, 6629–6634. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Ji, Y.; Zhong, C.; Liu, Y.; Tan, L.; Zhu, L. Fluorine-substituted benzothiadiazole-based hole transport materials for highly efficient planar perovskite solar cells with a FF exceeding 80%. Chem. Commun. 2017, 53, 8719–8722. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, S.Q.; Huo, L.J.; Zhang, M.J.; Hou, J.H. Molecular Design toward Highly Efficient Photovoltaic Polymers Based on Two-Dimensional Conjugated Benzodithiophene. Acc. Chem. Res. 2014, 47, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Jiao, X.C.; Zhou, M.; Zhang, S.Q.; Yao, H.F.; Zhao, W.C.; Xia, A.D.; Ade, H.; Hou, J.H. Manipulating Aggregation and Molecular Orientation in All-Polymer Photovoltaic Cells. Adv. Mater. 2015, 27, 6046–6054. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Ming, C.; Peng, L.; Jiajia, G.; Lars, K.; Licheng, S. Application of benzodithiophene based A–D–A structured materials in efficient perovskite solar cells and organic solar cells. Nano Energy 2016, 23, 40–49. [Google Scholar] [CrossRef]
- Yun, J.H.; Park, S.; Heo, J.H.; Lee, H.S.; Yoon, S.; Kang, J.; Im, S.H.; Kim, H.; Lee, W.; Kim, B.; et al. Enhancement of charge transport properties of small molecule semiconductors by controlling fluorine substitution and effects on photovoltaic properties of organic solar cells and perovskite solar cells. Chem. Sci. 2016, 7, 6649–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.S.; Hong, Z.R.; Chen, Q.; Chen, H.J.; Chang, W.H.; Yang, Y.; Song, T.B.; Yang, Y. Perovskite Solar Cells Employing Dopant-Free Organic Hole Transport Materials with Tunable Energy Levels. Adv. Mater. 2016, 28, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Sharmoukh, W.; Hassan, W.M.I.; Gros, P.C.; Allam, N.K. Design and synthesis of new Ru-complexes as potential photo-sensitizers: Experimental and TD-DFT insights. RSC Adv. 2016, 6, 69647–69657. [Google Scholar] [CrossRef]
- Mazzone, G.; Alberto, M.E.; De Simone, B.C.; Marino, T.; Russo, N. Can expanded bacteriochlorins act as photosensitizers in photodynamic therapy? Good news from density functional theory computations. Molecules 2016, 21, 288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.Z.; Song, P.; Su, R.Z.; Ma, F.C.; Yang, Y.H. Non-Fullerene Acceptor-Based Solar Cells: From Structural Design to Interface Charge Separation and Charge Transport. Polymers 2017, 9, 692. [Google Scholar] [CrossRef]
- Alberga, D.; Mangiatordi, G.F.; Labat, F.; Ciofini, I.; Nicolotti, O.; Lattanzi, G.; Adamo, C. Theoretical Investigation of Hole Transporter Materials for Energy Devices. J. Phys. Chem. C 2015, 119, 23890–23898. [Google Scholar] [CrossRef]
- Tian, L.; Feiwu, C. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar]
- Deng, W.Q.; Goddard, W.A. Predictions of Hole Mobilities in Oligoacene Organic Semiconductors from Quantum Mechanical Calculations. J. Phys. Chem. B 2004, 108, 8614–8621. [Google Scholar] [CrossRef]
- Berlin, Y.A.; Hutchison, G.R.; Rempala, P.; Ratner, M.A.; Michl, J. Charge Hopping in Molecular Wires as a Sequence of Electron-Transfer Reactions. J. Phys. Chem. A 2003, 107, 3970–3980. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Song, P.; Ma, F.; Sun, M. D-A-pi-A system: Light harvesting, charge transfer, and molecular designing. J. Phys. Chem. C 2017, 121, 12546–12561. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Ma, Y.; Ren, T.G.; Wang, L.; Zhang, J.L. Effects of π-conjugation on electrochemical properties within hole-transporting materials for perovskite solar cells from first principle and molecular dynamics. Org. Electron. 2017, 43, 96–104. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P. Design of two novel hole transport materials via replacing the core of spiro-OMeTAD with tetrathiafulvalene and tetraazafulvalene for application in perovskite solar cells. Sol. Energy 2018, 173, 132–138. [Google Scholar] [CrossRef]
- Yang, L.; Ren, A.M.; Feng, J.K.; Wang, J.F. Theoretical Investigation of Optical and Electronic Property Modulations of π-Conjugated Polymers Based on the Electron-Rich 3,6-Dimethoxy-fluorene Unit. J. Org. Chem. 2005, 70, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qi, Y.; Tang, Y.; Zheng, C.; Wan, Y.F.; Huang, W.; Chen, R.F. Controlling Intramolecular Conformation through Nonbonding Interaction for Soft-Conjugated Materials: Molecular Design and Optoelectronic Properties. J. Phys. Chem. Lett. 2016, 7, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 1985, 107, 6801–6806. [Google Scholar] [CrossRef]
- Jin, R.; Wang, K. Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells. Int. J. Mol. Sci. 2015, 16, 20326–20343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.Q.; Sun, L.; Huang, J.D.; Chai, S.; Wen, S.H.; Han, K.L. Quantitative prediction of charge mobilities of pi-stacked systems by first-principles simulation. Nat. Protoc. 2015, 10, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Sun, C.F.; Song, P.; Ma, F.C.; Yang, Y.H. Tuning the electron transport and accepting ability of dyes via introducing different π-conjugated bridges and acceptors for DSSCs. ChemPhysChem 2017, 18, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lin, X.C.; Mi, L.; Gao, N.; Song, P.; Ma, F.C. Characterizations of Efficient Charge Transfer and Photoelectric Performance in the Cosensitization of Solar Cells. Appl. Sci. 2018, 8, 1122. [Google Scholar] [CrossRef]
- Yin, J.; Chaitanya, K.; Ju, X.H. Theoretical investigations of charge carrier transport in organic semiconductors of naphthalene bisimides N-substituted with alkoxyphenyl groups. Can. J. Chem. 2015, 93, 740–748. [Google Scholar] [CrossRef]
- Cornil, J.; Brédas, J.; Zaumseil, J.; Sirringhaus, H. Ambipolar Transport in Organic Conjugated Materials. Adv. Mater. 2007, 19, 1791–1799. [Google Scholar] [CrossRef]
- Li, H.; Zheng, R.; Shi, Q. Theoretical Study of Charge Carrier Transport in Organic Semiconductors of Tetrathiafulvalene Derivatives. J. Phys. Chem. C 2012, 116, 11886–11894. [Google Scholar] [CrossRef]
- Lan, Y.K.; Huang, C.I. A Theoretical Study of the Charge Transfer Behavior of the Highly Regioregular Poly-3-hexylthiophene in the Ordered State. J. Phys. Chem. B 2008, 112, 14857–14862. [Google Scholar] [CrossRef] [PubMed]
- Coropceanu, V.; Cornil, J.; Demetrio, A.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.-L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nan, G.; Yang, X.; Peng, Q.; Li, Q.; Shuai, Z. Computational methods for design of organic materials with high charge mobility. Chem. Soc. Rev. 2010, 39, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Ming-Yu, K.; Hsing-Yin, C.; Ito, C. Cyanation: Providing a Three-in-One Advantage for the Design of n-Type Organic Field-Effect Transistors. Chem. Eur. J. 2007, 13, 4750–4758. [Google Scholar]







{kind=link}
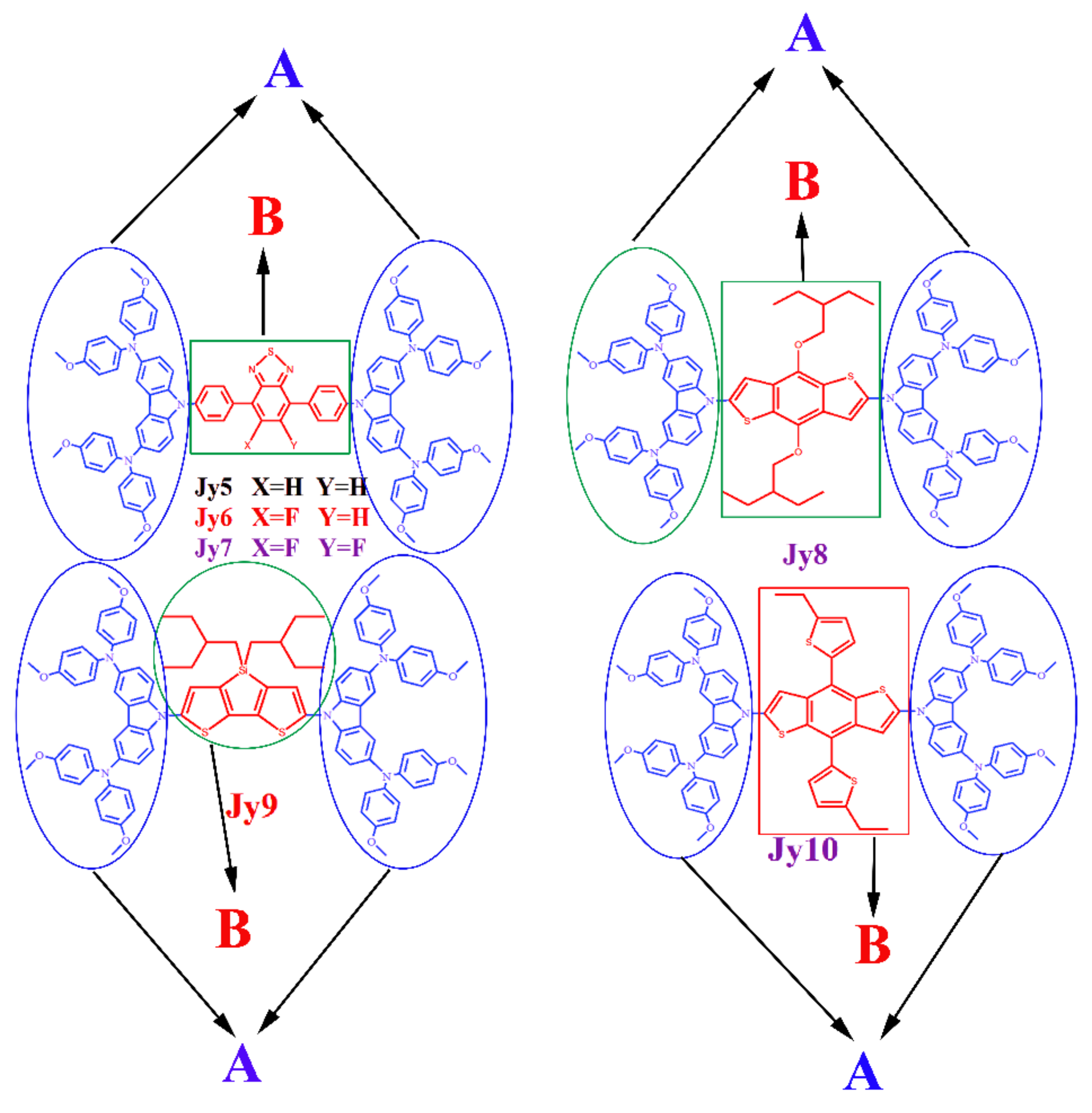{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | HOMO (eV) | LUMO (eV) | ΔH-L (eV) |
|---|---|---|---|
| Jy5 | −4.49 | −2.55 | 1.94 |
| Jy6 | −4.49 | −2.59 | 1.90 |
| Jy7 | −4.50 | −2.62 | 1.88 |
| Jy8 | −4.53 | −1.46 | 3.07 |
| Jy9 | −4.53 | −1.71 | 2.82 |
| Jy10 | −4.52 | −1.70 | 2.82 |
| Molecules | Jy5 | Jy6 | Jy7 | Jy8 | Jy9 | Jy10 |
|---|---|---|---|---|---|---|
| IP | 4.40 | 4.39 | 4.41 | 4.45 | 4.37 | 4.44 |
| EA | 2.64 | 2.68 | 2.70 | 1.66 | 1.96 | 1.98 |
| η | 0.88 | 0.86 | 0.86 | 1.40 | 1.21 | 1.23 |
| Molecules | Jy5 | Jy6 | Jy7 | Jy8 | Jy9 | Jy10 |
|---|---|---|---|---|---|---|
| λh | 0.14 | 0.15 | 0.15 | 0.11 | 0.17 | 0.11 |
| λe | 0.34 | 0.35 | 0.36 | 0.39 | 0.39 | 0.42 |
| Molecules | State | Energy (eV) | λ (nm) | Transition MO | f |
|---|---|---|---|---|---|
| Jy5 | S1 | 3.07 | 404.04 | (0.51528) H→L | 0.9012 |
| S2 | 3.32 | 373.98 | (0.67782) H-1→L | 0.0028 | |
| S3 | 3.58 | 346.08 | (0.45233) H→L | 0.0917 | |
| S4 | 3.64 | 340.52 | (0.35359) H-1→L+2 | 0.0612 | |
| S5 | 3.64 | 340.33 | (0.38410) H→L+2 | 0.0378 | |
| S6 | 3.72 | 333.54 | (0.68793) H-2→L | 0.0093 | |
| Jy6 | S1 | 3.05 | 405.84 | (0.50908) H→L | 0.8423 |
| S2 | 3.27 | 379.98 | (0.63588) H-1→L | 0.0021 | |
| S3 | 3.57 | 346.78 | (0.39596) H→L | 0.1722 | |
| S4 | 3.64 | 340.41 | (0.50734) H-1→L+2 | 0.0807 | |
| S5 | 3.64 | 340.32 | (0.40186) H→L+3 | 0.0124 | |
| S6 | 3.66 | 338.53 | (0.68005) H-3→L | 0.0122 | |
| Jy7 | S1 | 3.09 | 400.73 | (0.57491) H→L | 0.7346 |
| S2 | 3.25 | 381.79 | (0.66434) H-1→L | 0.0018 | |
| S3 | 3.62 | 342.21 | (0.36991) H-12→L | 0.2870 | |
| S4 | 3.64 | 340.56 | (0.44441) H→L+2 | 0.0320 | |
| S5 | 3.65 | 339.77 | (0.50438) H-2→L | 0.0104 | |
| S6 | 3.65 | 339.54 | (0.39980) H-1→L+3 | 0.0415 |
| Molecules | State | Energy (eV) | λ (nm) | Transition MO | f |
|---|---|---|---|---|---|
| Jy8 | S1 | 3.61 | 343.01 | (0.44085) H→L | 1.4740 |
| S2 | 3.66 | 339.00 | (0.47233) H→L+1 | 0.0001 | |
| S3 | 3.68 | 336.98 | (0.38674) H→L+2 | 0.2549 | |
| S4 | 3.94 | 314.85 | (0.37167) H-3→L+2 | 0.0020 | |
| S5 | 3.94 | 314.65 | (0.33494) H-2→L+1 | 0.4762 | |
| S6 | 3.96 | 313.06 | (0.59462) H-1→L | 0.0045 | |
| Jy9 | S1 | 3.54 | 349.88 | (0.58077) H-4→L | 0.8324 |
| S2 | 3.68 | 337.11 | (0.40054) H→L+2 | 0.0379 | |
| S3 | 3.68 | 336.70 | (0.43639) H-1→L+1 | 0.0335 | |
| S4 | 3.83 | 324.09 | (0.56164) H-1→L | 0.0074 | |
| S5 | 3.92 | 315.94 | (0.45648) H→L | 0.1017 | |
| S6 | 3.95 | 314.17 | (0.42169) H-2→L+1 | 0.0989 | |
| Jy10 | S1 | 3.49 | 355.46 | (0.52453) H→L | 1.3031 |
| S2 | 3.65 | 339.49 | (0.47197) H→L+1 | 0.0032 | |
| S3 | 3.66 | 338.81 | (0.46339) H→L+2 | 0.0447 | |
| S4 | 3.79 | 326.63 | (0.61019) H-1→L | 0.0001 | |
| S5 | 3.84 | 322.60 | (0.50900) H-4→L | 0.3618 | |
| S6 | 3.94 | 314.70 | (0.44812) H-2→L+1 | 0.2635 |
| Molecules | Jy5 | Jy6 | Jy7 |
|---|---|---|---|
| Vh (eV) | 0.00016 | 0.00141 | 0.01001 |
| λh (eV) | 0.14 | 0.15 | 0.15 |
| r (Å) | 4.65 | 7.94 | 7.19 |
| kh (s−1) | 2.96 × 108 | 2.01 × 1010 | 1.01 × 1012 |
| μh (cm2/(V·s) | 1.24 × 10−5 | 2.45 × 10−3 | 0.1010 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Lin, X.; Cao, X.; Song, P.; Ma, F.; Li, Y. Molecular Controlling the Transport Properties for Benzothiadiazole-Based Hole Transport Materials. Appl. Sci. 2018, 8, 1461. https://doi.org/10.3390/app8091461
Liu Q, Lin X, Cao X, Song P, Ma F, Li Y. Molecular Controlling the Transport Properties for Benzothiadiazole-Based Hole Transport Materials. Applied Sciences. 2018; 8(9):1461. https://doi.org/10.3390/app8091461
Chicago/Turabian StyleLiu, Qian, Xiaochen Lin, Xinlan Cao, Peng Song, Fengcai Ma, and Yuanzuo Li. 2018. "Molecular Controlling the Transport Properties for Benzothiadiazole-Based Hole Transport Materials" Applied Sciences 8, no. 9: 1461. https://doi.org/10.3390/app8091461
APA StyleLiu, Q., Lin, X., Cao, X., Song, P., Ma, F., & Li, Y. (2018). Molecular Controlling the Transport Properties for Benzothiadiazole-Based Hole Transport Materials. Applied Sciences, 8(9), 1461. https://doi.org/10.3390/app8091461