Study of a Miniaturizable System for Optical Sensing Application to Human Cells

 , ,
, ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Labeling
2.3. Fluorescence and Confocal Microscopy
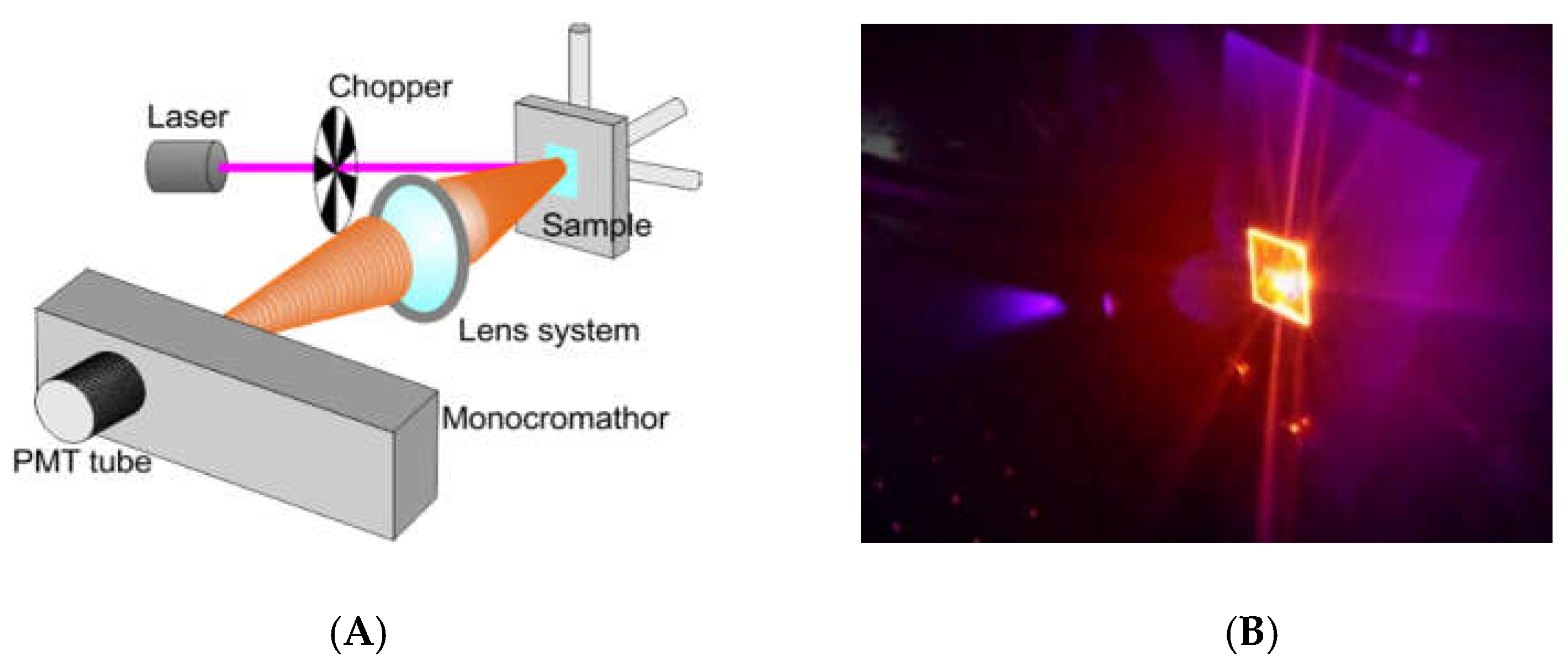
2.4. Optical Measurement for Bio-Sensing Applications
2.5. MTT Assay on HDF and LoVo
3. Results
3.1. Method Characterization
3.1.1. HDFs Fluorescent Labeling
3.1.2. HDFs Confocal Microscopy
3.1.3. Photomultiplier-based System for Labeled Fibroblast Analysis
3.1.4. HDFs Vitality
3.2. Tumor Cell Fluorescent Time-Lapse Analysis
3.2.1. LoVo Uptake of Ru(bpy)32+
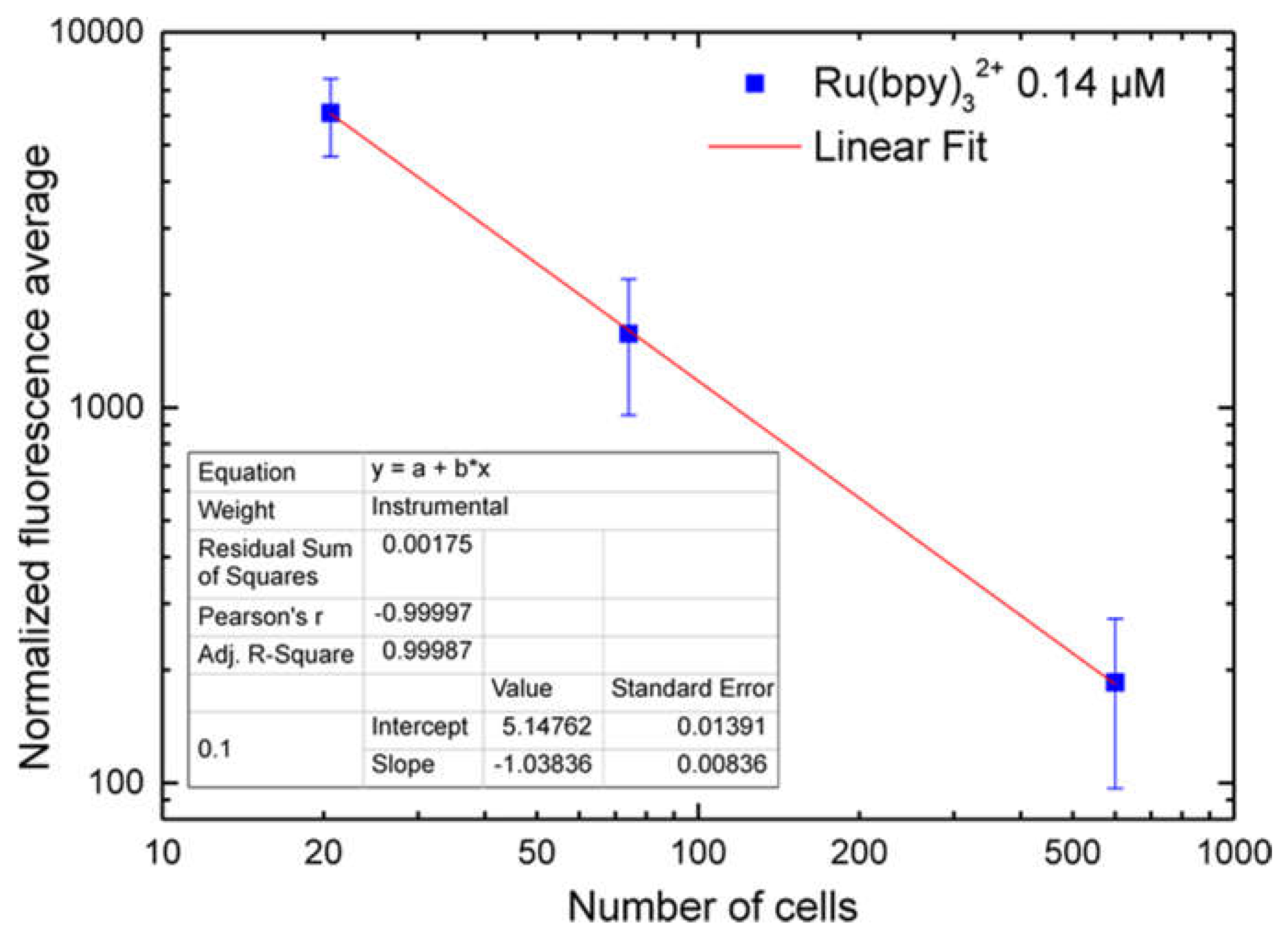
3.2.2. LoVo Fluorescence Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strickland, L.; von Dassow, G.; Ellenberg, J.; Foe, V.; Lenart, P.; Burgess, D. Light Microscopy of Echinoderm Embryos. Met. Cell Biol. 2004, 74, 371–409. [Google Scholar]
- Sud, D.; Mycek, M.A. Calibration and validation of an optical sensor for intracellular oxygen measurements. J. Biomed. Opt. 2009, 14, 020506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.D.; Searson, P.C. Live-Cell Imaging of Invasion and Intravasation in an Artificial Microvessel Platform. Cancer Res. 2014, 74, 4937–4945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abulateefeh, S.R.; Spain, S.G.; Thurecht, K.J.; Aylott, J.W.; Chan, W.C.; Garnett, M.C.; Alexander, C. Enhanced uptake of nanoparticle drug carriers via a thermoresponsive shell enhances cytotoxicity in a cancer cell line. Biomat. Sci. 2013, 1, 434–442. [Google Scholar] [CrossRef]
- Bastiat, G.; Pritz, C.O.; Roider, C.; Fouchet, F.; Lignières, E.; Jesacherd, A.; Glueckert, R.; Ritsch-Mart, M.; Schrott-Fischer, A.; Saulnier, P.; et al. A new tool to ensure the fluorescent dye labeling stability of nanocarriers: A real challenge for fluorescence imaging. J. Control. Release 2013, 170, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, M.; Alexander, L.; Sanchez-Martin, R.M. Cellular uptake of fluorescent labeled biotin-streptavidin microspheres. J. Fluoresc. 2008, 18, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Meijering, E.; Smal, I.; Dzyubachyk, O.; Olivo-Marin, J. Time-Lapse Imaging; Elsevier Academic Press: Cambridge, MA, USA, 2008; Chapter 15; pp. 401–440. [Google Scholar]
- Celis, J.E. Cell Biology, 3rd ed.; Elsevier Academic Press: Cambridge, MA, USA, 2006; ISBN 9780121647308. [Google Scholar]
- Herman, B. Fluorescence Microscopy, 2nd ed.; Bios Scientific Pub.: Milton Park, UK, 1998; ISBN 0387915516. [Google Scholar]
- Lakowicz, J.R. Radiative Decay Engineering: Biophysical and Biomedical Applications. Anal. Biochem. 2001, 298, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, M.F.; Sciuto, E.L.; Lombardo, S.A.; Busacca, A.C.; Petralia, S.; Conoci, S.; Libertino, S. Si Photomultipliers for Bio-Sensing Applications. IEEE Select. Top. Quantum Electron. 2016, 22, 3. [Google Scholar]
- Sciuto, E.L.; Santangelo, M.F.; Villaggio, G.; Sinatra, F.; Bongiorno, C.; Nicotra, G.; Libertino, S. Photo-physical characterization of fluorophore Ru(bpy)32+ for optical biosensing applications. SBSR 2015, 6, 67–71. [Google Scholar]
- Cannone, F.; Chirico, G.; Diaspro, A. Two-photon interactions at single fluorescent molecule level. J. Biomed. Opt. 2003, 8, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Medepalli, K.; Alphenaar, B.W.; Keynton, R.S.; Sethu, P. A new technique for reversible permeabilization of live cells for intracellular delivery of quantum dots. Nanotechnology 2013, 24, 205101. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Van Houten, J.; Watts, R.J. Temperature dependence of the photophysical and photochemical properties of the tris(2,2′-bipyridyl)ruthenium(II) ion in aqueous solution. J. Am. Chem. Soc. 1976, 98, 4853–4858. [Google Scholar] [CrossRef]
- Gill, M.R.; Thomas, J.A. Ruthenium(II) polypyridyl complexes and DNA—From structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef] [PubMed]
- Tsui, W.; Chung, L.; Wong, M.M.; Tsang, W.; Lo, H.; Liu, Y.; Leung, C.; Ma, D.; Chiu, S.; Wong, C. Luminescent Ruthenium(II) Complex Bearing Bipyridine and N-Heterocyclic Carbene-based C∧N∧C Pincer Ligand for Live-Cell Imaging of Endocytosis. Sci. Rep. 2015, 5, 9070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puckett, C.A.; Barton, J.K. Methods to Explore Cellular Uptake of Ruthenium Complexes. J. Am. Chem. Soc. 2007, 129, 46–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, S.J.; Filby, M.H.; Whitehouse, A.; Wilson, A.J. Cellular uptake of highly-functionalized ruthenium(II) tris-bipyridine protein-surface mimetics. Bioorg. Med. Chem. Lett. 2012, 22, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Gyenge, E.B.; Darphin, X.; Wirth, A.; Pieles, U.; Walt, H.; Bredell, M.; Maake, C. Uptake and fate of surface modified silica nanoparticles in head and neck squamous cell carcinoma. J. Nanobiotech. 2011, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatzschneider, U.; Niesel, J.; Ott, I.; Gust, R.; Alborzinia, H.; Wçlfl, S. Cellular Uptake, Cytotoxicity, and Metabolic Profiling of Human Cancer Cells Treated with Ruthenium(II) Polypyridyl Complexes [Ru (bpy)2 (N_N)]Cl2 with N_N=bpy, phen, dpq, dppz, and dppn**. Chem. Med. Chem. 2008, 3, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Federico, C.; Gil, L.; Bruno, F.; D’Amico, A.G.; D’Agata, V.; Saccone, S. Phosphorylated nucleolar Tau protein is related to the neuronal in vitro differentiation. Gene 2018, 664, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Petralia, S.; Sciuto, E.L.; Messina, M.A.; Mirabella, S.; Scandurra, A.; Priolo, F.; Conoci, S. Miniaturized and Multi-Purpose Electrochemical Sensing Device based on thin Ni Oxides. Sens. Act. B Chem. 2018, 263, 10–19. [Google Scholar] [CrossRef]
- Petralia, S.; Sciuto, E.L.; Santangelo, M.F.; Messina, M.A.; Libertino, S.; Conoci, S. Sulphide Species Optical Monitoring by Miniaturized Silicon Photomultiplier. Sensors 2018, 18, 727. [Google Scholar] [CrossRef] [PubMed]
- Petralia, S.; Cosentino, T.; Sinatra, F.; Favetta, M.; Fiorenza, P.; Bongiorno, C.; Sciuto, E.L.; Conoci, S.; Libertino, S. Silicon Nitride Surfaces as Active Substrate for Electrical DNA Biosensors. Sens. Act. B Chem. 2017, 252, 492–502. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciuto, E.L.; Villaggio, G.; Santangelo, M.F.; Laudani, S.; Federico, C.; Saccone, S.; Sinatra, F.; Libertino, S. Study of a Miniaturizable System for Optical Sensing Application to Human Cells. Appl. Sci. 2019, 9, 975. https://doi.org/10.3390/app9050975
Sciuto EL, Villaggio G, Santangelo MF, Laudani S, Federico C, Saccone S, Sinatra F, Libertino S. Study of a Miniaturizable System for Optical Sensing Application to Human Cells. Applied Sciences. 2019; 9(5):975. https://doi.org/10.3390/app9050975
Chicago/Turabian StyleSciuto, Emanuele Luigi, Giusy Villaggio, Maria Francesca Santangelo, Samuele Laudani, Concetta Federico, Salvatore Saccone, Fulvia Sinatra, and Sebania Libertino. 2019. "Study of a Miniaturizable System for Optical Sensing Application to Human Cells" Applied Sciences 9, no. 5: 975. https://doi.org/10.3390/app9050975
APA StyleSciuto, E. L., Villaggio, G., Santangelo, M. F., Laudani, S., Federico, C., Saccone, S., Sinatra, F., & Libertino, S. (2019). Study of a Miniaturizable System for Optical Sensing Application to Human Cells. Applied Sciences, 9(5), 975. https://doi.org/10.3390/app9050975