Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets Downloadand Bioinformatic and Statistical Data Analysis
2.2. Machine Learning Data Analysis
3. Results
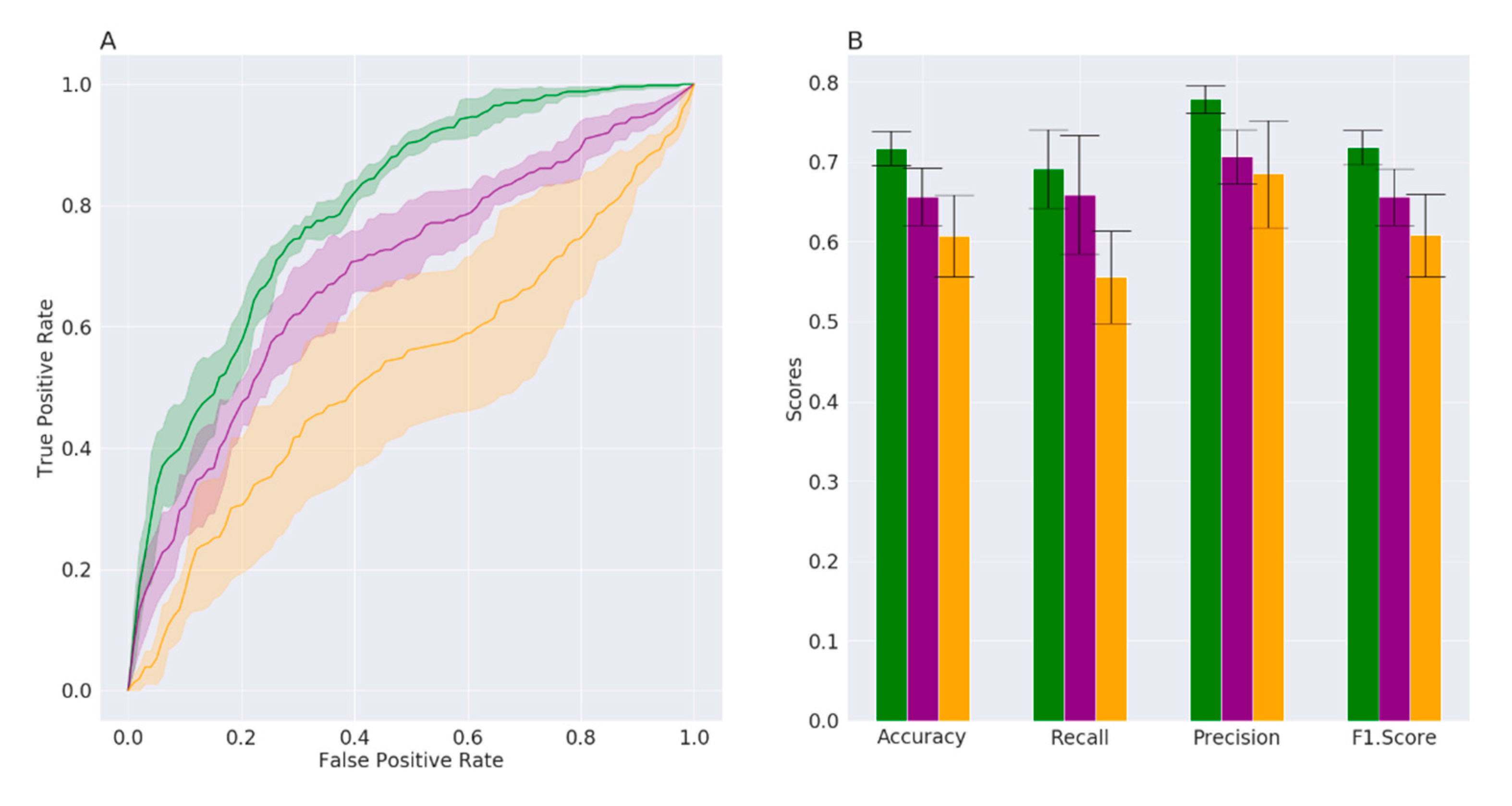
3.1. Datasets Description and Supervised Machine Learning Approach
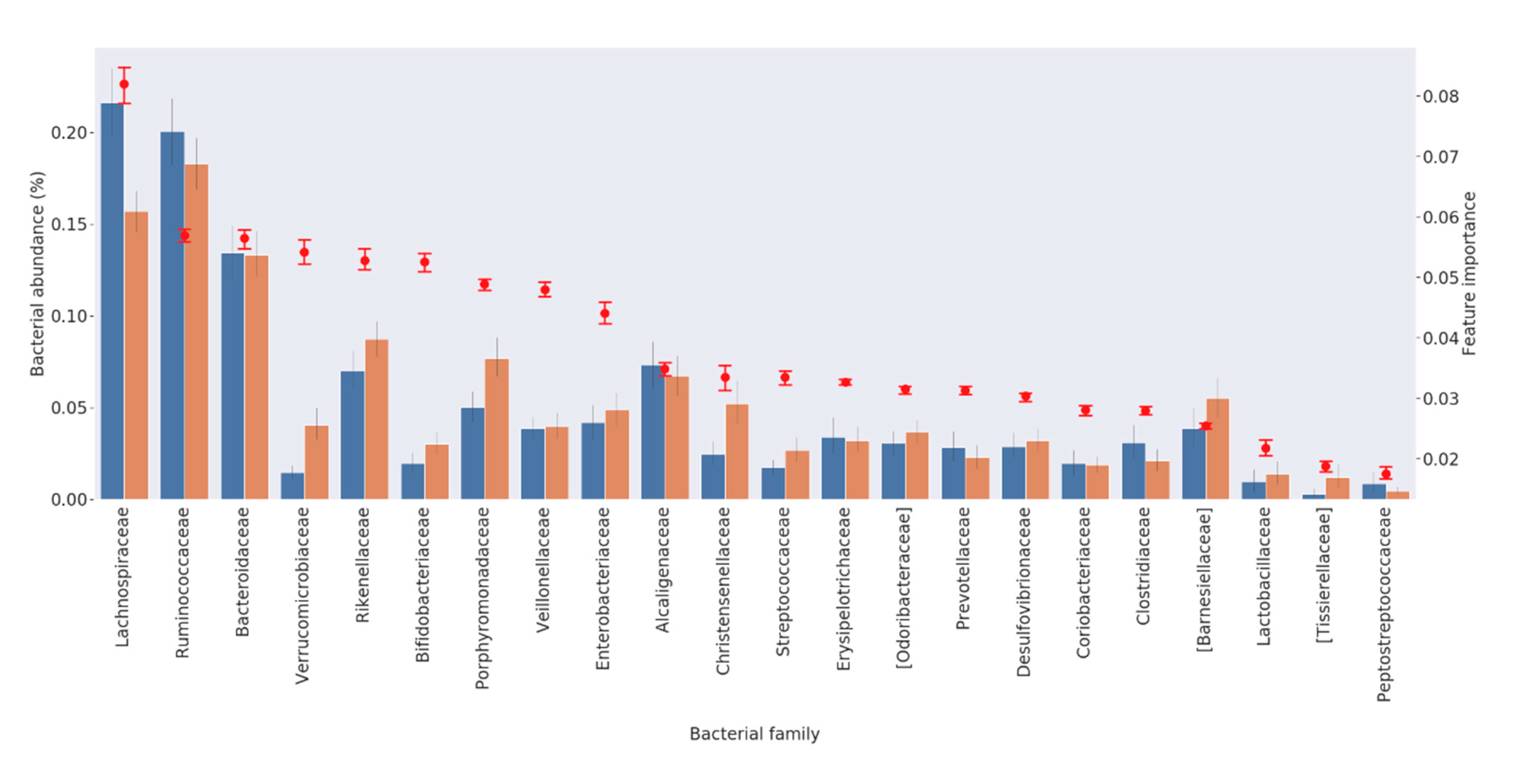
3.2. Feature Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grochowska, M.; Laskus, T.; Radkowski, M. Gut Microbiota in Neurological Disorders. Arch. Immunol. Ther. Exp. (Warsz.) 2019, 67, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Haikal, C.; Chen, Q.; Li, J. Microbiome changes: An indicator of Parkinson’s disease? Transl. Neurodegener. 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, Y.; El-Hadidi, M.; Admard, J.; Wassouf, Z.; Schulze-Hentrich, J.M.; Kohlhofer, U.; Quintanilla-Martinez, L.; Huson, D.; Riess, O.; Casadei, N. Enriched environmental conditions modify the gut microbiome composition and fecal markers of inflammation in parkinson’s disease. Front. Neurosci. 2019, 13, 1032. [Google Scholar] [CrossRef] [Green Version]
- Boertien, J.M.; Pereira, P.A.B.; Aho, V.T.E.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Parkinsons Dis. 2019, 9, S297–S312. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Bove, F.; Gabrielli, M.; Petracca, M.; Zocco, M.A.; Ragazzoni, E.; Barbaro, F.; Piano, C.; Fortuna, S.; Tortora, A.; et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov. Disord. 2013, 28, 1241–1249. [Google Scholar] [CrossRef]
- Fitzgerald, E.; Murphy, S.; Martinson, H.A. Alpha-synuclein pathology and the role of the microbiota in Parkinson’s disease. Front. Neurosci. 2019, 13, 369. [Google Scholar] [CrossRef]
- Pietrucci, D.; Cerroni, R.; Unida, V.; Farcomeni, A.; Pierantozzi, M.; Mercuri, N.B.; Biocca, S.; Stefani, A.; Desideri, A. Dysbiosis of gut microbiota in a selected population of Parkinson’s patients. Park. Relat. Disord. 2019, 65, 124–130. [Google Scholar] [CrossRef]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, F.; Künstner, A.; Müller, S.H.; Künzel, S.; Zeuner, K.E.; Margraf, N.G.; Deuschl, G.; Baines, J.F.; Kuhlenbäumer, G. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017, 1667, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, P.; Chen, Z.; Sui, X.; Xie, X.; Zhang, J. Alteration of the fecal microbiota in North-Eastern Han Chinese population with sporadic Parkinson’s disease. Neurosci. Lett. 2019, 707, 134297. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of gut microbiota in patients with parkinson’s disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2019, 34, 396–405. [Google Scholar] [CrossRef]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39–52. [Google Scholar] [CrossRef]
- Lin, A.; Zheng, W.; He, Y.; Tang, W.; Wei, X.; He, R.; Huang, W.; Su, Y.; Huang, Y.; Zhou, H.; et al. Gut microbiota in patients with Parkinson’s disease in southern China. Park. Relat. Disord. 2018, 53, 82–88. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A machine learning approach for identifying gene biomarkers guiding the treatment of breast cancer. Front. Genet. 2019, 10, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavakiotis, I.; Tsave, O.; Salifoglou, A.; Maglaveras, N.; Vlahavas, I.; Chouvarda, I. Machine Learning and Data Mining Methods in Diabetes Research. Comput. Struct. Biotechnol. J. 2017, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.F.; Reps, J.; Kai, J.; Garibaldi, J.M.; Qureshi, N. Can Machine-learning improve cardiovascular risk prediction using routine clinical data? PLoS ONE 2017, 12, e0174944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, W.A.; Xu, Z.; Knight, R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014, 588, 4223–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; Desantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Samborska, I.A.; Alexandrov, V.; Sieczko, L.; Kornatowska, B.; Goltsev, V.; Cetner, M.D.; Kalaji, H.M. Artificial neural networks and their application in biological and agricultural research. J. NanoPhotoBioSci. 2014, 2, 14–30. [Google Scholar]
- Yang, Z.R. Biological applications of support vector machines. Brief. Bioinform. 2004, 5, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | PD Samples | HC Samples | Sample Transport | DNA Extraction | 16S Region | Nationality |
|---|---|---|---|---|---|---|
| Method | ||||||
| [2] | 34 | 31 | BD Gaspak | FastDNA Spin Kit for Soil | V4 | United States |
| [10] | 65 | 68 | NR | PSP Spin Stool Kit | V3-V4 | Finland |
| [9] | 116 | 82 | Stabilizer PSP | PSP Spin Stool Kit | V3-V4 | Italy |
| [13] | 206 | 133 | Ambient temp | Earth microbiome project protocol | V4 | United States |
| [12] | 22 | 34 | Stabilizer PSP | PSP Spin Stool Kit | V3-V4 | Germany |
| [11] | 29 | 26 | Immediate freezing | Custom Protocol Hopfner | V4 | Russia |
| Bacterial Family | Ranking of Importance | Higher (−) or Lower (+) Abundance in PD Patients from RF Algorithm | References in the Literature Reporting Overabundance in PD Patients | References in the Literature Reporting Lower Abundance in PD Patients |
|---|---|---|---|---|
| Lachnospiraceae | 1 | − | [14] | [2,9,10,11,13,15,16,17] |
| Ruminococcaceae | 2 | − | [2,14] | [13,15] |
| Bacteroidaceae | 3 | − | [2] | [16] |
| Verrucomicrobiaceae | 4 | + | [2,12,13,17] | |
| Rikenellaceae | 5 | + | [19] | |
| Bifidobacteriaceae | 6 | + | [13,16,17,19] | [2,18] |
| Porphyromonadaceae | 7 | + | [17] | |
| Veillonellaceae | 8 | + | ||
| Enterobacteriaceae | 9 | + | [9,15,17] | |
| Alcaligenaceae | 10 | − | ||
| Streptococcaceae | 11 | + | [19] | |
| Christensenellaceae | 12 | + | [13,16,17] | |
| Erysipelotrichaceae | 13 | + | [19] | |
| [Odoribacteraceae] | 14 | + | ||
| Prevotellaceae | 15 | − | [10,16,17] | |
| Desulfovibrionaceae | 16 | + | [14,19] | |
| Coriobacteriaceae | 17 | − | [17] | |
| Clostridiaceae | 18 | − | [2,10,16] | [19] |
| [Barnesiellaceae] | 19 | + | ||
| Lactobacillaceae | 20 | + | [9,10,11,16] | |
| [Tissierellaceae] | 21 | + | ||
| Peptostreptococcaceae | 22 | − | [16,17] | [14,15] |
| Methanobacteriaceae | 23 | + | [19] | |
| [Mogibacteriaceae] | 24 | − | ||
| [Paraprevotellaceae] | 25 | + | ||
| Turicibacteraceae | 26 | − | ||
| Pseudomonadaceae | 27 | + | ||
| Victivallaceae | 28 | − | ||
| Campylobacteraceae | 29 | + | ||
| Synergistaceae | 30 | + | ||
| Pasteurellaceae | 31 | − | [14] | [10,13,19] |
| Corynebacteriaceae | 32 | + | ||
| S24-7 | 33 | − | ||
| Enterococcaceae | 34 | + | [9,11,15,17] | |
| Actinomycetaceae | 35 | + | ||
| Moraxellaceae | 36 | − | ||
| Burkholderiaceae | 37 | − | ||
| Comamonadaceae | 38 | + | ||
| Alcanivoracaceae | 39 | − | ||
| Oxalobacteraceae | 40 | − | ||
| Propionibacteriaceae | 41 | − | ||
| Xanthomonadaceae | 42 | − | ||
| Rhodobacteraceae | 43 | − | ||
| Fusobacteriaceae | 44 | + | ||
| Staphylococcaceae | 45 | − | ||
| Caulobacteraceae | 46 | + | ||
| Caldicoprobacteraceae | 47 | − | ||
| Succinivibrionaceae | 48 | + | ||
| Peptococcaceae | 49 | − | ||
| Flavobacteriaceae | 50 | + | ||
| [Weeksellaceae] | 51 | + | ||
| Aeromonadaceae | 52 | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrucci, D.; Teofani, A.; Unida, V.; Cerroni, R.; Biocca, S.; Stefani, A.; Desideri, A. Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach. Brain Sci. 2020, 10, 242. https://doi.org/10.3390/brainsci10040242
Pietrucci D, Teofani A, Unida V, Cerroni R, Biocca S, Stefani A, Desideri A. Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach. Brain Sciences. 2020; 10(4):242. https://doi.org/10.3390/brainsci10040242
Chicago/Turabian StylePietrucci, Daniele, Adelaide Teofani, Valeria Unida, Rocco Cerroni, Silvia Biocca, Alessandro Stefani, and Alessandro Desideri. 2020. "Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach" Brain Sciences 10, no. 4: 242. https://doi.org/10.3390/brainsci10040242
APA StylePietrucci, D., Teofani, A., Unida, V., Cerroni, R., Biocca, S., Stefani, A., & Desideri, A. (2020). Can Gut Microbiota Be a Good Predictor for Parkinson’s Disease? A Machine Learning Approach. Brain Sciences, 10(4), 242. https://doi.org/10.3390/brainsci10040242