Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion

 , , , ,
, , , , 
Abstract
:1. Introduction
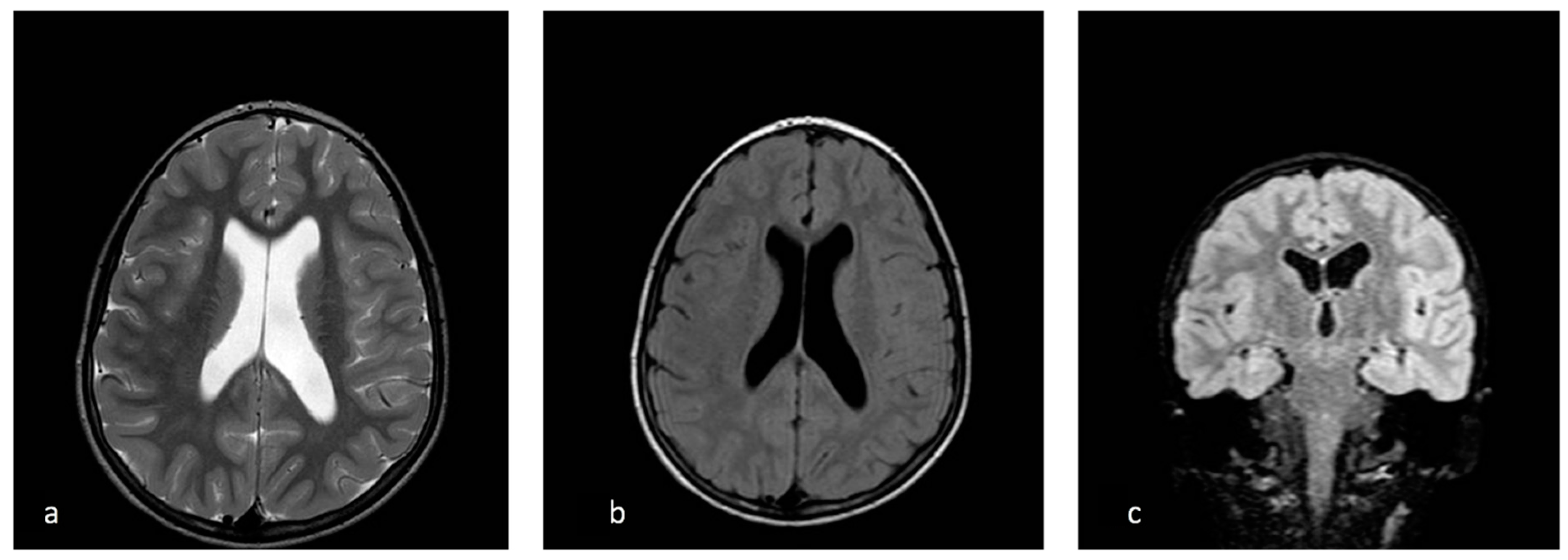
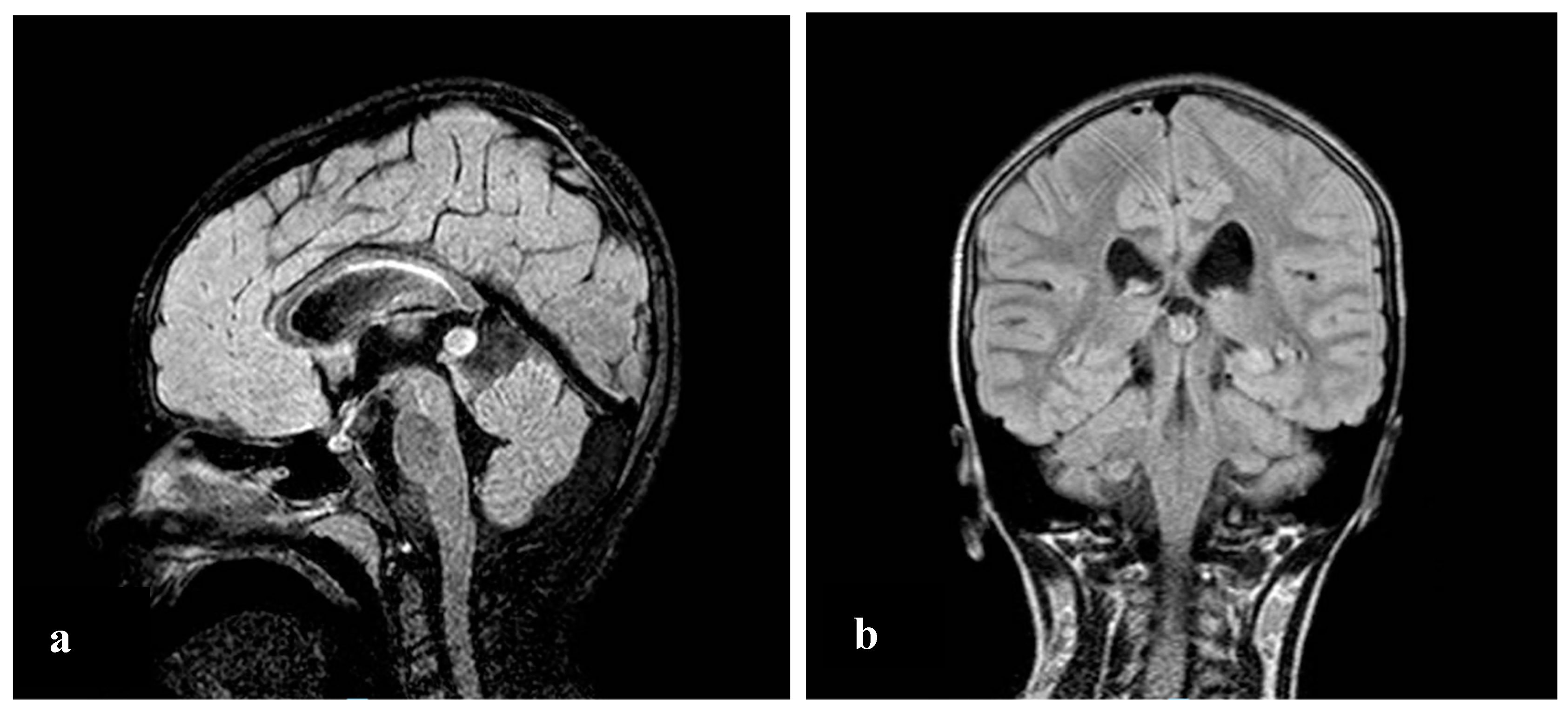
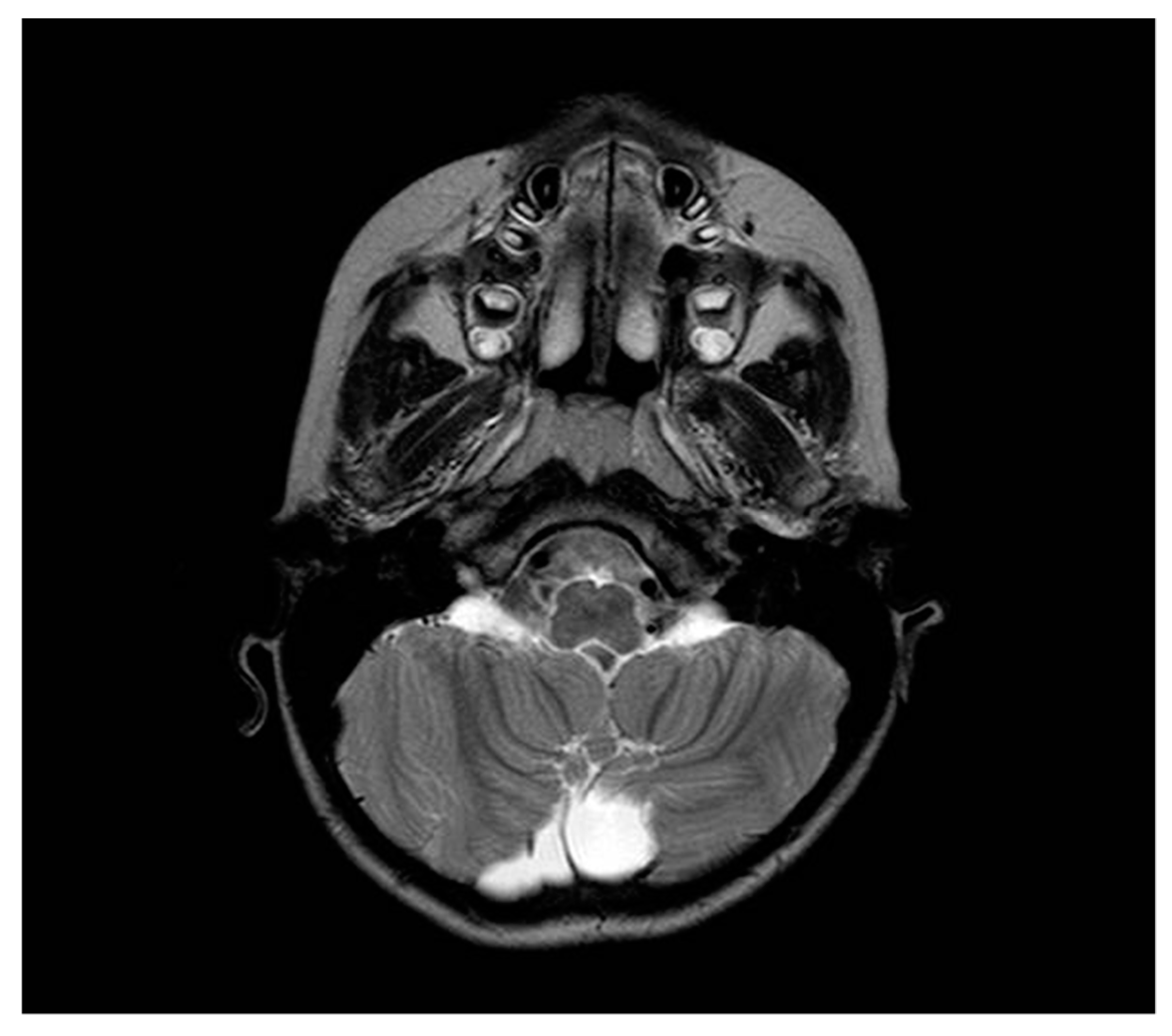
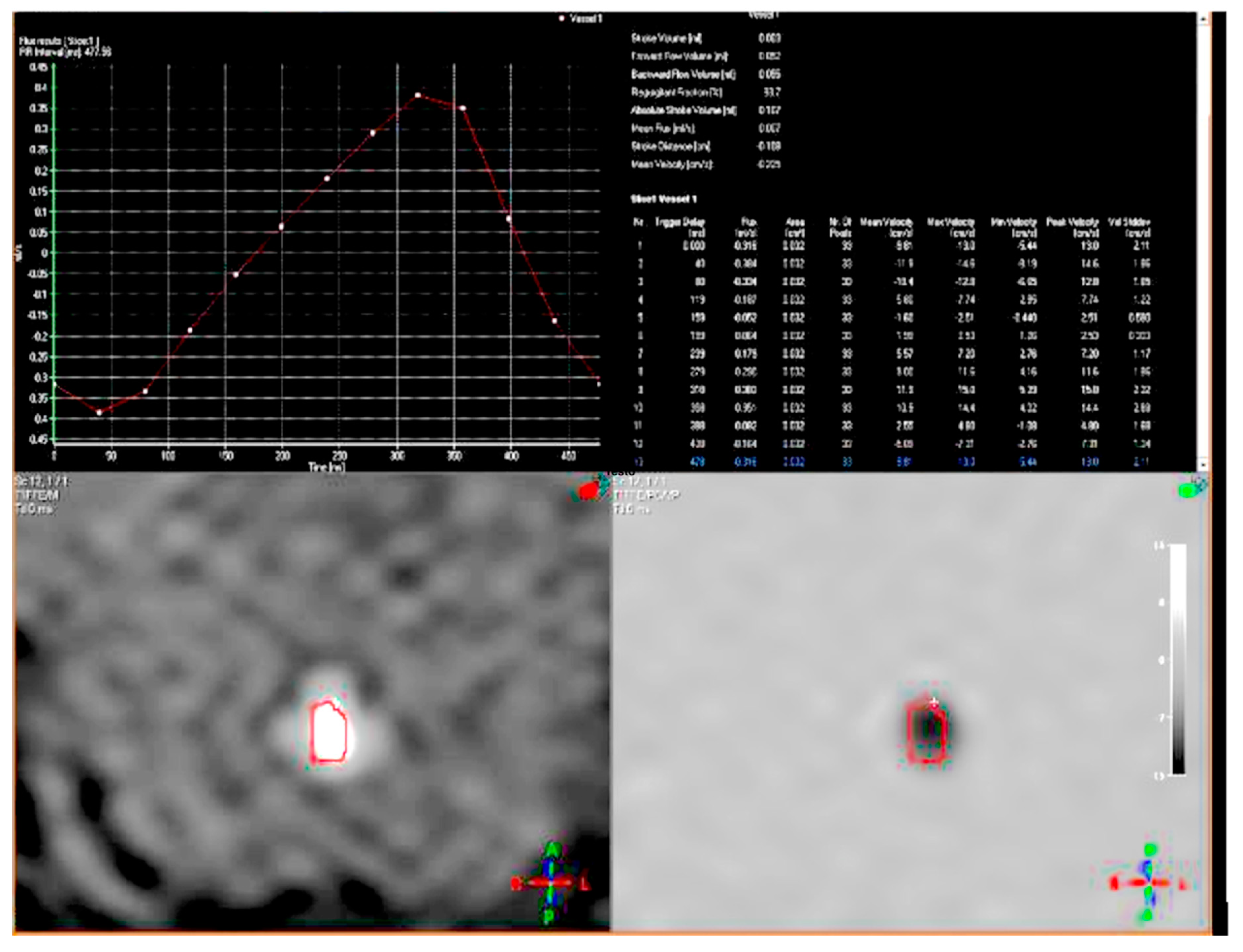
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- García-Santiago, F.A.; Martínez-Glez, V.; Santos, F.; García-Miñaur, S.; Mansilla, E.; Meneses, A.G.; Rosell, J.; Granero, Á.P.; Vallespín, E.; Fernández, L.; et al. Analysis of invdupdel(8p) rearrangement: Clinical, cytogenetic and molecular characterization. Am. J. Med. Genet. A 2015, 167, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Floridia, G.; Piantanida, M.; Minelli, A.; Dellavecchia, C.; Bonaglia, C.; Rossi, E.; Gimelli, G.; Croci, G.; Franchi, F.; Gilgenkrantz, S.; et al. The same molecular mechanism at the maternal meiosis I produces mono- and dicentric 8p duplications. Am. J. Hum. Genet. 1996, 58, 785–796. [Google Scholar]
- Ciccone, R.; Mattina, T.; Giorda, R.; Bonaglia, M.C.; Rocchi, M.; Pramparo, T.; Zuffardi, O. Inversion polymorphisms and non-contiguous terminal deletions: The cause and the (unpredicted) effect of our genome architecture. J. Med. Genet. 2006, 43, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am. J. Hum. Genet. 2001, 68, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, O.; Kurosawa, K.; Ida, T.; Harada, N.; Kondoh, T.; Miyake, N.; Yoshiura, K.; Kishino, T.; Ohta, T.; Niikawa, N.; et al. Molecular characterization of inv dup del(8p): Analysis of five cases. Am. J. Med. Genet. A 2004, 128, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Giglio, S.; Calvari, V.; Gregato, G.; Gimelli, G.; Camanini, S.; Giorda, R.; Ragusa, A.; Guerneri, S.; Selicorni, A.; Stumm, M.; et al. Heterozygous Submicroscopic Inversions Involving Olfactory Receptor–Gene Clusters Mediate the Recurrent t(4;8)(p16;p23) Translocation. Am. J. Hum. Genet. 2002, 71, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Piccione, M.; Salzano, E.; Vecchio, D.; Ferrara, D.; Malacarne, M.; Pierluigi, M.; Ferrara, I.; Corsello, G. 4p16.1-p15.31 duplication and 4p terminal deletion in a 3-years old Chinese girl: Array-CGH, genotype-phenotype and neurological characterization. Eur. J. Paediatr. Neurol. 2015, 19, 477–483. [Google Scholar] [CrossRef]
- Vermeesch, J.; Thoelen, R.; Salden, I.; Raes, M.; Matthijs, G.; Fryns, J. Mosaicism del(8p)/inv dup(8p) in a dysmorphic female infant: A mosaic formed by a meiotic error at the 8p OR gene and an independent terminal deletion event. J. Med. Genet. 2003, 40, e93. [Google Scholar] [CrossRef] [Green Version]
- de Die-Smulders, C.E.; Engelen, J.J.; Schrander-Stumpel, C.T.; Govaerts, L.C.; de Vries, B.; Vles, J.S.; Wagemans, A.; Schijns-Fleuren, S.; Gillessen-Kaesbach, G.; Fryns, J.P. Inversion duplication of the short arm of chromosome 8: Clinical data on seven patients and review of the literature. Am. J. Med. Genet. 1995, 59, 369–374. [Google Scholar] [CrossRef]
- Devriendt, K.; Matthijs, G.; Van Dael, R.; Gewillig, M.; Eyskens, B.; Hjalgrim, H.; Dolmer, B.; McGaughran, J.; Bröndum-Nielsen, K.; Marynen, P.; et al. Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am. J. Hum. Genet. 1999, 64, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Ergün, M.A.; Kula, S.; Karaer, K.; Perçin, E.F. A case with de novo inv dup del(8p) associated with dextrocardia and corpus callosum agenesis. Pediatr. Int. 2010, 52, 845–846. [Google Scholar] [CrossRef]
- Masuda, K.; Nomura, Y.; Yoshinaga, M.; Nakamura, M.; Matsuda, Y.; Oku, S.; Miyata, K. Inverted duplication/deletion of the short arm of chromosome 8 in two patients with tetralogy of Fallot. Pediatr. Int. 2002, 44, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Hand, M.; Gray, C.; Glew, G.; Tsuchiya, K.D. Mild phenotype in a patient with mosaic del(8p)/inv dup del(8p). Am. J. Med. Genet. A 2010, 152, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Buysse, K.; Antonacci, F.; Callewaert, B.; Loeys, B.; Fränkel, U.; Siu, V.; Mortier, G.; Speleman, F.; Menten, B. Unusual 8p inverted duplication deletion with telomere capture from 8q. Eur. J. Med. Genet. 2009, 52, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.S.; Siu, V.M. Molecular cytogenetic characterization of a derivative chromosome 8 with an inverted duplication of 8p21.3-->p23.3 and a rearranged duplication of 8q24.13-->qter. Am. J. Med. Genet. 2001, 102, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Roy, S.; Kumar, G. An Interesting and Unique Case of 8p23.3p23.1 Deletion and 8p23.1p11.1 Interstitial Duplication Syndrome. J. Pediatr. Genet. 2018, 7, 125–129. [Google Scholar] [CrossRef]
- Cooke, S.L.; Northup, J.K.; Champaige, N.L.; Zinser, W.; Edwards, P.A.W.; Lockhart, L.H.; Velagaleti, G.V.N. Molecular cytogenetic characterization of a unique and complex de novo 8p rearrangement. Am. J. Med. Genet. Part A 2008, 146, 1166–1172. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Engelen, J.J.M.; Ghesquiere, S.; Alofs, M.; Saatci, C.; Dundar, M. Fluorescence in situ hybridization and single nucleotide polymorphism of a new case with inv dup del(8p). Genet. Couns. 2009, 20, 333–340. [Google Scholar] [PubMed]
- Fisch, G.S.; Davis, R.; Youngblom, J.; Gregg, J. Genotype-phenotype association studies of chromosome 8p inverted duplication deletion syndrome. Behav. Genet. 2011, 41, 373–380. [Google Scholar] [CrossRef]
- Knijnenburg, J.; Uytdewilligen, M.E.W.; van Hassel, D.A.C.M.; Oostenbrink, R.; Eussen, B.H.J.; de Klein, A.; Brooks, A.S.; van Zutven, L.J.C.M. Postzygotic telomere capture causes segmental UPD, duplication and deletion of chromosome 8p in a patient with intellectual disability and obesity. Eur. J. Med. Genet. 2017, 60, 445–450. [Google Scholar] [CrossRef]
- Piro, E.; Consiglio, V.; Agrifoglio, M.; Sireci, F.; Ballacchino, A.; Salvago, P.; Martines, F.; Graziano, F.; Busè, M.; Sanfilippo, C.; et al. Diagnosis and follow-up of complex congenital malformations/mental retardation (MRA/MR). Acta Med. Mediterr. 2013, 29, 321–325. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Karyotype/CGH-Arrays | FISH | Phenotype |
|---|---|---|---|
| Fan et al. | karyotype of 46,XY,add(8)(p23) 46,XY,der(8)(qter→q24.13::p21.3→p23.3::p23.3→qter) | + | Global developmental delay. Marked hypotonia, weak low cry. Bitemporal low set ears, upslanting palpable fissures, wide nasal bridge, right cleft lip, micrognathia, excess nuchal skin, hypoplastic and widely spaced nipples. Left testis in the inguinal canal. Atrial septal defect, membranous ventricular septal defect, patent ductus arteriosus with a parachute mitral valve. Right pelvic dysplastic kidney and left hydronephrosis. Partial agenesis of the corpus callosum, communicating hydrocephalus, Dandy Walker malformation, intramedullary cord defect. |
| Masuda et al. | der(8) (qter→p23.1::p23.1→p12:) | + | Severe motor delay and mental impairment. Hypotonia. Prominent forehead, posteriorly angulated ears, broad nose with depressed nasal bridge, wide mouth, high-arched palate and downward slanting eyes. Tetralogy of Fallot (TOF). Agenesis of the corpus callosum. |
| Vermeesch et al. | 46,XX,del(8)(p23.3) inv dup(8)(p21.1p23.2)/46,XX,del(8)(p21.1) | + | Delayed psychomotor development. Axial hypotonia. Upward slanting palpebral fissures, synophys, and left preauricular tag, low set thumbs with hypotrophic thenars, bilateral clinodactyly of the fifth fingers. Linear areas of depigmentation with bordering areas of hyperpigmentation on the lumbar and presacral region and on both legs. Feeding problems with gastro-esophageal reflux [8]. |
| Ciccone et al. | 46,XX,psu dic(8)(p23.2)/46,XX,del(8)(p23.1) | + | Severe mental impairment. Asymmetrical face with the left eye lower than the right, left palpebral ptosis, dental malocclusion, zygomatic arch hypoplasia, low set ears, and a short neck with webbing. Kyphoscoliosis, globous abdomen, short upper and lower limbs, premature grey hair. |
| Cooke et al. | 46,XX,der(8)dir dup(8)(p21p23.1) del(8)(p23.1p- ter).ish der(8)dir dup(8)(p21p23.1)del(8)(p23.1pter) (wcp8þ,pter -) | + | Global developmental delays. No meaningful speech. Poor auditory attention, impulsiveness and decreased attention span. Upward slanting palpebral fissures, epicanthal folds, low columella with hypoplastic alae nasi, smooth philtrum, thin vermilion to the upper lip, high arched palate, bilateral clinodactyly. Partial complex seizures. Recurrent upper and lower respiratory tract infections. Mild degree of brain atrophy and evidence of a Dandy–Walker variant in the posterior fossa. |
| Caglayan et al. | Del 8p23.1: 6.99 Mb;Dup 8p11.2→8p23.1: 31.51 Mb | nr | Severe cognitive delay. Microcephaly, frontal bossing, malformed ears, thin vermilion of upper lip, abnormal maxilla and mandible, strabismus, coloboma. Corpus callosum agenesis. |
| Buysse et al. | 46,XY,der(8)(qter→q24.13::p21.3→p23.3::p23.3→qter) Del 8p23.1l: 6.9 Mb; Dup 8p22: 3.4 Mb;Dup 8qter→24.13: 20.9 Mb | + | Global developmental delay. Hypertelorism, intermittent strabismus of the left eye, hetero-chromia iridis of the right eye, upslanting palpebral fissures, blue sclerae, slight retrognathia, ears posteriorly rotated with a preauricular tag on the left side. Intergluteal hairy dimple. Supravalvular pulmonary stenosis. Bilateral decreased vision with astigmatism and hypermetropia. |
| Hand et al. | Del 8p23.1: 6.8 Mb;Mosaic Del 8p21.2: 1.7Mb; Mosaic Dup p21.2→p23.1:11Mb | - | Cognitive, speech and motor delays. Hypotonia. Bilateral single palmar creases, no clinodactyly. Skin pigmentary abnormalities (faint lines of hyperpigmentation on the backs of the both legs). No evidence of facial dysmorphisms. Cheerful disposition, eager to please. |
| Ergun et al. | Del 8p23.1: 6.71 Mb;Dup 8p11.2!8p23.1: 29.26 Mb | nr | Absent nasal bone and clenched left hand. Enlarged thickened heart walls along with polyvalvular dysplasia. Dilatation of the main pulmonary artery and branches. History of necrotizing enterocolitis. Agenesis of the corpus callosum, enlarged third ventricle and cerebellar hypoplasia. |
| Fisch et al. |
| nr |
|
| Garcìa-Santiago et al. |
| + |
|
| Knijnenburg et al. | 46 XY | + | Moderate intellectual disability. Flat occiput, epicanthal folds, downturned corners of the mouth, broad based nose, broad hands with tapering fingers and mild 2-3 toe syndactyly. Atrial septal defect. Obesity. Occasionally aggressive outbursts. |
| Kumar et al. | 6.7- Mb deletion on chromosome 8p23.3p23.1 and a 31-Mb interstitial duplication on chromosome 8p23.1p11.1. | nr | Global developmental delay. Generalized hypotonia. Broad forehead, low set ears, thick lips, prominent philtrum. Harrison sulcus. History of generalized seizures. Large doubly committed ventricular septal defect (VSD) with left to right shunt and severe hyperkinetic pulmonary artery hypertension. Colpocephaly with complete absence of corpus callosum, prominent ventricles. |
| Our patient | 46, XX, der(8)del(8)(p23.1)invdup(p12p23.1) | + | Developmental and speech delay. No meaningful sentences. Hypotonia. Hypothyroidism. Prominent forehead, arched eyebrow, thin nose with rounded tip and anteverse nostrils, flat filter, thin down-turned lips, slight micrognathia, low-set posteriorly rotated ears. Single palmar crease on the right hand and bilateral IV-V fingers clinodactyly. Hypertrichosis, previous sacrococcygeal fistula sign. Extra-rotation of the lower limbs, varus position of both the knees, flat feet. Bilateral cutaneous dimples on both elbows and knees, shield chest, inverted nipples, winged shoulder blades. Emotiveness, impulsiveness, decreased attention span. Dilatation of lateral ventricles, pineal gland’s small ectasia, moderate cystic cisterna magna’s ectasia, retrocerebellar cystic ectasia. Global chorio-retinic dystrophia, pale papilla with clear boundaries, peri-papillar pigmentary ring. Sialorrhea and extravelic palatin tonsils, ogival palate, type C tympanogram with absent stapedial reflex on the left. |
| Article | Year | No. of Patients | Sex | Dysmorphisms | Intellectual Disabilities/Behavioural Disorders | Brain MRI Anomalies | Congenital Heart Defects | Abdominal Anomalies | Skeletal Anomalies |
|---|---|---|---|---|---|---|---|---|---|
| Fan et al. | 2001 | 1 | M | + | + | + | + | + | − |
| Masuda et al. | 2002 | 2 | 1F/1M | + | + | + | + | − | − |
| Vermeesch et al. | 2003 | 1 | F | + | + | nr | − | − | − |
| Ciccone et al. | 2006 | 1 | F | + | + | nr | − | − | + |
| Cooke et al. | 2008 | 1 | F | + | + | + | − | − | + |
| Caglayan et al. | 2009 | 1 | ? | + | + | + | − | − | − |
| Buysse et al. | 2009 | 1 | F | + | + | nr | + | − | − |
| Hand et al. | 2010 | 1 | F | − | + | nr | + | − | − |
| Ergun et al. | 2010 | 1 | F | + | + | + | + | − | − |
| Fisch et al. | 2011 | 4 | 2F/2M | + | + | nr | − | − | − |
| Garcìa−Santiago et al. | 2014 | 7 | 4F/3M | + | + | + | + | − | + |
| Knijnenburg et al. | 2017 | 1 | M | + | + | nr | + | − | + |
| Kumar et al. | 2018 | 1 | M | + | + | + | + | − | + |
| Our patient | 2020 | 1 | F | + | + | + | − | − | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Bianco, M.; Vecchio, D.; Timpanaro, T.A.; Arena, A.; Macchiaiolo, M.; Bartuli, A.; Sciuto, L.; Presti, S.; Sciuto, S.; Sapuppo, A.; et al. Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion. Brain Sci. 2020, 10, 451. https://doi.org/10.3390/brainsci10070451
Lo Bianco M, Vecchio D, Timpanaro TA, Arena A, Macchiaiolo M, Bartuli A, Sciuto L, Presti S, Sciuto S, Sapuppo A, et al. Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion. Brain Sciences. 2020; 10(7):451. https://doi.org/10.3390/brainsci10070451
Chicago/Turabian StyleLo Bianco, Manuela, Davide Vecchio, Tiziana A. Timpanaro, Alessia Arena, Marina Macchiaiolo, Andrea Bartuli, Laura Sciuto, Santiago Presti, Sarah Sciuto, Annamaria Sapuppo, and et al. 2020. "Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion" Brain Sciences 10, no. 7: 451. https://doi.org/10.3390/brainsci10070451
APA StyleLo Bianco, M., Vecchio, D., Timpanaro, T. A., Arena, A., Macchiaiolo, M., Bartuli, A., Sciuto, L., Presti, S., Sciuto, S., Sapuppo, A., Fiumara, A., Marino, L., Messina, G., & Pavone, P. (2020). Deciphering the Invdupdel(8p) Genotype–Phenotype Correlation: Our Opinion. Brain Sciences, 10(7), 451. https://doi.org/10.3390/brainsci10070451