Levetiracetam Reduced the Basal Excitability of the Dentate Gyrus without Restoring Impaired Synaptic Plasticity in Rats with Temporal Lobe Epilepsy


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Pilocarpine-Induced Status Epilepticus
2.2. Monitoring of Spontaneous Behavioral Seizures
2.3. Levetiracetam Treatment
2.4. Electrophysiological Recordings
2.4.1. Input/Output Curves
2.4.2. Paired-Pulse Depression and Facilitation
2.4.3. Long-Term Potentiation
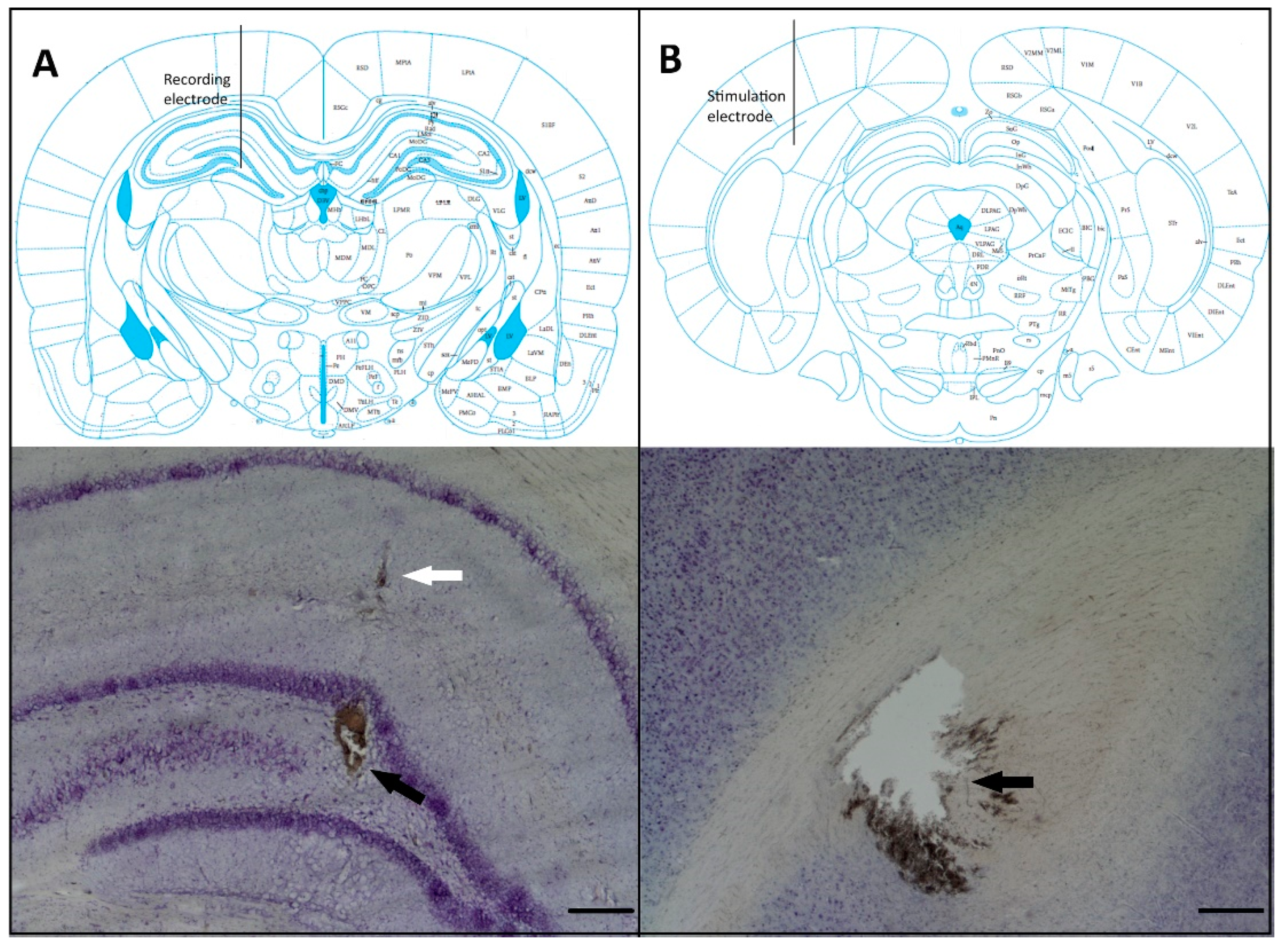
2.5. Histological Verification
2.6. Statistical Analysis
3. Results
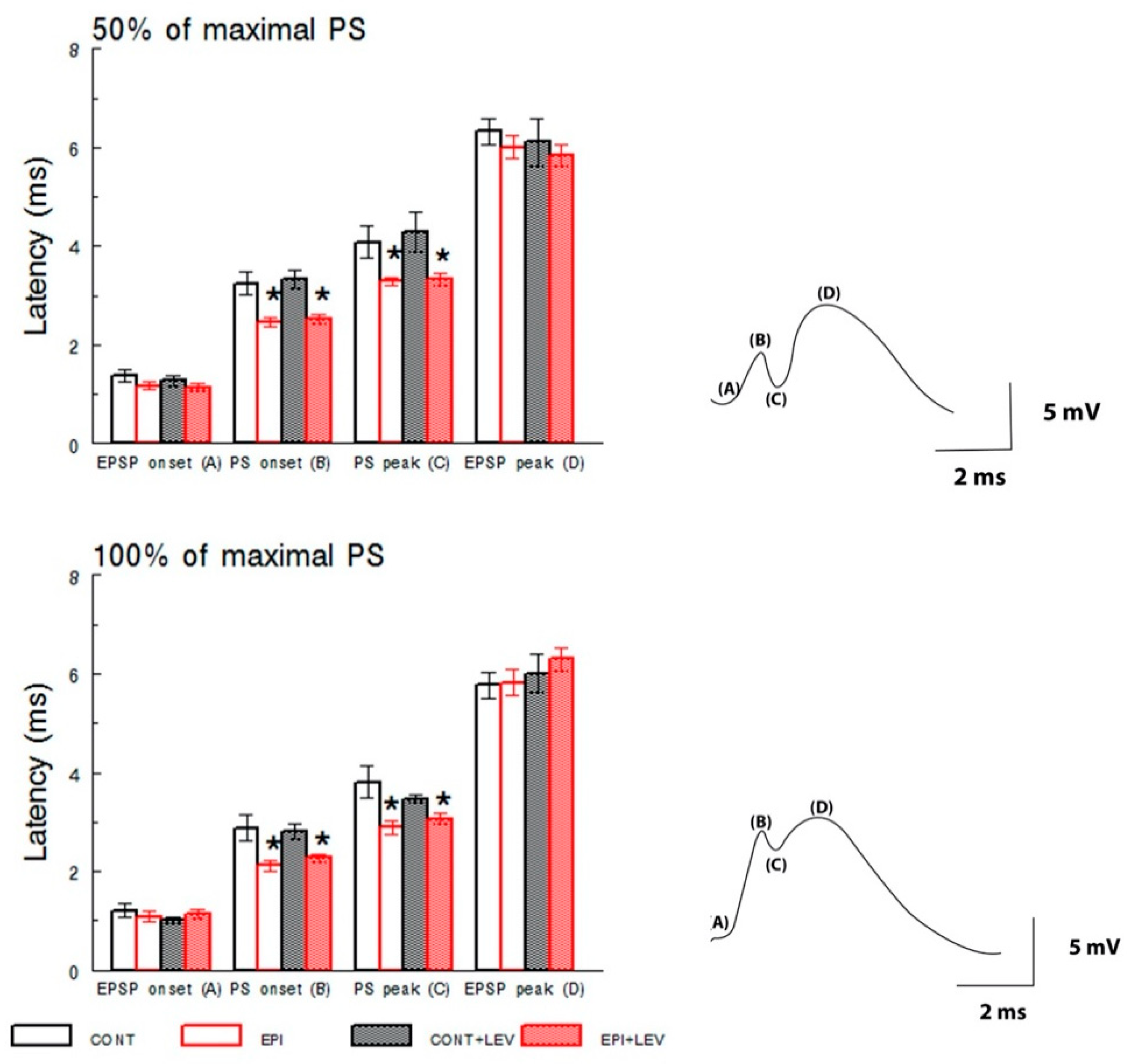
3.1. Basal Excitatory Synaptic Transmission: Input/Output Curve
3.2. Short-Term Plasticity: Paired-Pulse Facilitation and Depression
3.3. Long-Term Plasticity
4. Discussion
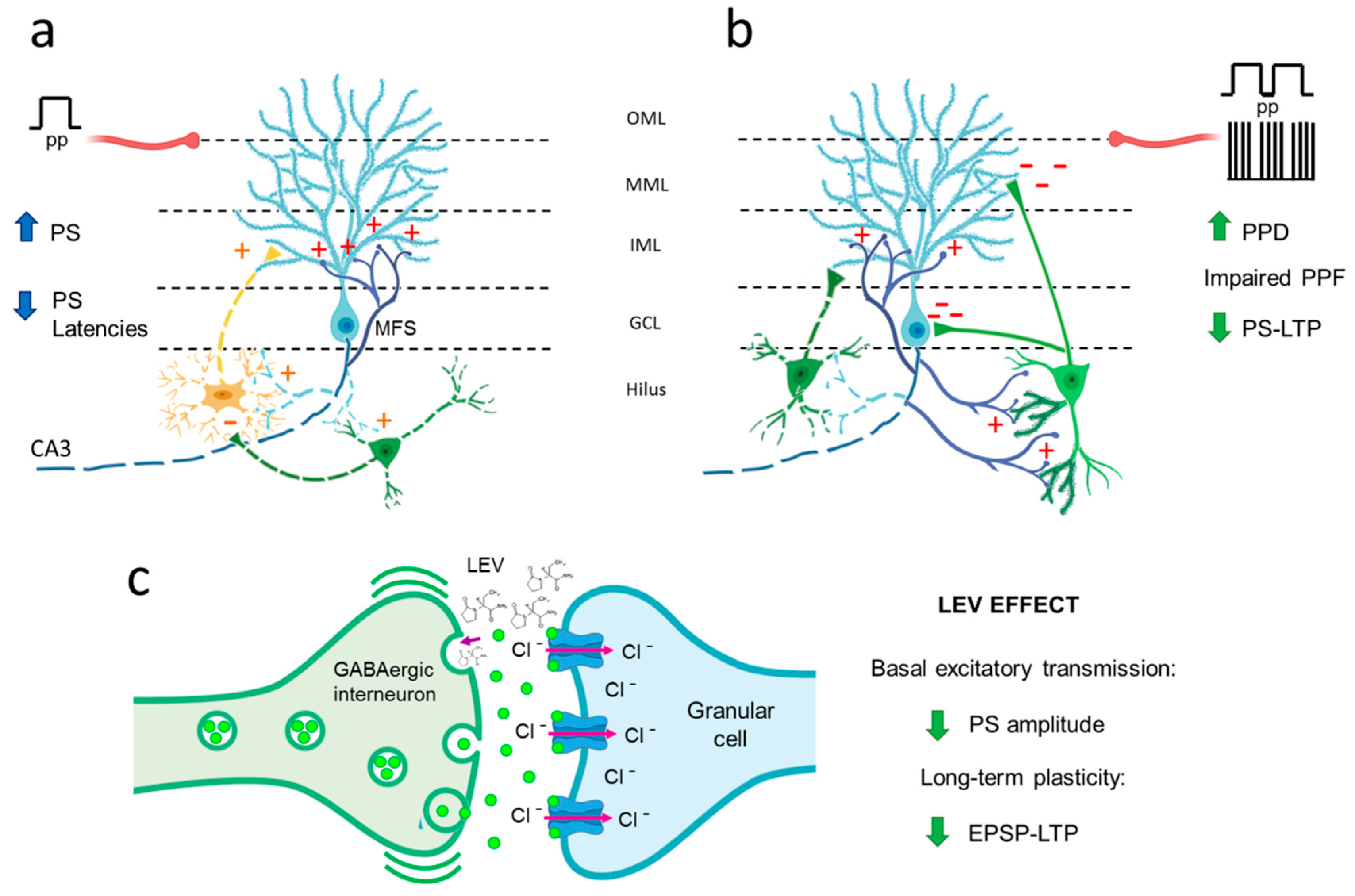
4.1. Hyperexcitability Caused by TLE in the DG Is Attenuated by LEV Treatment
4.2. TLE Caused Alterations in Short-Term (Facilitation/Depression) Synaptic Plasticity, and LEV Did Not Reverse Them
4.3. TLE Reduced PS-LTP, and LEV Did Not Correct This Alteration
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| CONT | control |
| DG | dentate gyrus |
| EPI | epileptic |
| EPSP | excitatory postsynaptic potential |
| GABA | γ-aminobutyric acid |
| I/O | input/output |
| IPI | interpulse interval |
| LEV | levetiracetam |
| LTP | long-term potentiation |
| PP | paired-pulse |
| PPD | paired-pulse depression |
| PPF | paired-pulse facilitation |
| PS | population spike |
| RM | repeated-measures |
| S.E.M. | standard error of the mean |
| SE | status epilepticus |
| S–N–K | Student–Newman–Keuls |
| SV2A | synaptic vesicle protein 2A |
| TLE | temporal lobe epilepsy |
References
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E. Pathophysiological mechanisms of seizures and epilepsy: A primer. In Epilepsy: Mechanisms, Models, and Translational Perspectives; Rho, J.M., Sankar, R., Stafstrom, C.E., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 3–17. [Google Scholar]
- Engel, J., Jr. Mesial temporal lobe epilepsy: What have we learned? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2001, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Semah, F.; Picot, M.C.; Adam, C.; Broglin, D.; Arzimanoglou, A.; Bazin, B.; Cavalcanti, D.; Baulac, M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Zenteno, J.F.; Hernandez-Ronquillo, L. A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res. Treat. 2012, 2012, 630853. [Google Scholar] [CrossRef]
- Engel, J., Jr. Introduction to temporal lobe epilepsy. Epilepsy Res. 1996, 26, 141–150. [Google Scholar] [CrossRef]
- Blair, R.D. Temporal lobe epilepsy semiology. Epilepsy Res. Treat. 2012, 2012, 751510. [Google Scholar] [CrossRef]
- Sheng, J.; Liu, S.; Qin, H.; Li, B.; Zhang, X. Drug-Resistant Epilepsy and Surgery. Curr. Neuropharmacol. 2018, 16, 17–28. [Google Scholar] [CrossRef]
- Zhao, F.; Kang, H.; You, L.; Rastogi, P.; Venkatesh, D.; Chandra, M. Neuropsychological deficits in temporal lobe epilepsy: A comprehensive review. Ann. Indian Acad. Neurol. 2014, 17, 374–382. [Google Scholar] [CrossRef]
- Jokeit, H.; Ebner, A. Long term effects of refractory temporal lobe epilepsy on cognitive abilities: A cross sectional study. J. Neurol. Neurosurg. Psychiatry 1999, 67, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhao, C.W.; Long, Z.; Xiao, B.; Feng, L. Anatomy Based Networks and Topology Alteration in Seizure-Related Cognitive Outcomes. Front. Neuroanat. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Hermann, B.; Seidenberg, M.; Lee, E.J.; Chan, F.; Rutecki, P. Cognitive phenotypes in temporal lobe epilepsy. J. Int. Neuropsychol. Soc. 2007, 13, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, B.P.; Seidenberg, M.; Dow, C.; Jones, J.; Rutecki, P.; Bhattacharya, A.; Bell, B. Cognitive prognosis in chronic temporal lobe epilepsy. Ann. Neurol. 2006, 60, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M. Synaptic plasticity and learning and memory: 15 years of progress. Neurobiol. Learn. Mem. 1998, 70, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryukov, K.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. Neuroreport 2016, 27, 1191–1195. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Zubareva, O.E.; Kovalenko, A.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status Epilepticus Impairs Synaptic Plasticity in Rat Hippocampus and Is Followed by Changes in Expression of NMDA Receptors. Biochem. Biokhimiia 2017, 82, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, C.M.; Mello, L.E. Synaptic plasticity of the CA3 commissural projection in epileptic rats: An in vivo electrophysiological study. Eur. J. Neurosci. 2007, 25, 3071–3079. [Google Scholar] [CrossRef]
- Ge, Y.X.; Tian, X.Z.; Lin, Y.Y.; Liu, X.Y. Chronic treatment with levetiracetam reverses deficits in hippocampal LTP in vivo in experimental temporal lobe epilepsy rats. Neurosci. Lett. 2016, 628, 194–200. [Google Scholar] [CrossRef]
- Carpenter-Hyland, E.; Bichler, E.K.; Smith, M.; Sloviter, R.S.; Benveniste, M. Epileptic pilocarpine-treated rats exhibit aberrant hippocampal EPSP-spike potentiation but retain long-term potentiation. Physiol. Rep. 2017, 5, e13490. [Google Scholar] [CrossRef]
- Levesque, M.; Behr, C.; Avoli, M. The anti-ictogenic effects of levetiracetam are mirrored by interictal spiking and high-frequency oscillation changes in a model of temporal lobe epilepsy. Seizure 2015, 25, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.M.; Gillard, M.; Klitgaard, H. Targeting SV2A for Discovery of Antiepileptic Drugs. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Bethesda: Rockville, MA, USA, 2012. [Google Scholar]
- Gronborg, M.; Pavlos, N.J.; Brunk, I.; Chua, J.J.; Munster-Wandowski, A.; Riedel, D.; Ahnert-Hilger, G.; Urlaub, H.; Jahn, R. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Torreblanca, J.G.; Garcia-Cruz, M.E.; Sanchez-Cruz, I.; Gomez-Gonzalez, B.; Juarez-Mendez, S.; Gomez-Lira, G. Analysis of Differential Expression of Synaptic Vesicle Protein 2A in the Adult Rat Brain. Neuroscience 2019, 419, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Crowder, K.M.; Gunther, J.M.; Jones, T.A.; Hale, B.D.; Zhang, H.Z.; Peterson, M.R.; Scheller, R.H.; Chavkin, C.; Bajjalieh, S.M. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. USA 1999, 96, 15268–15273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custer, K.L.; Austin, N.S.; Sullivan, J.M.; Bajjalieh, S.M. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Nowack, A.; Kensel-Hammes, P.; Gardner, R.G.; Bajjalieh, S.M. Cotrafficking of SV2 and synaptotagmin at the synapse. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 5569–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.F.; Weisenfeld, A.; Rothman, S.M. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia 2007, 48, 1861–1869. [Google Scholar] [CrossRef]
- Meehan, A.L.; Yang, X.; McAdams, B.D.; Yuan, L.; Rothman, S.M. A new mechanism for antiepileptic drug action: Vesicular entry may mediate the effects of levetiracetam. J. Neurophysiol. 2011, 106, 1227–1239. [Google Scholar] [CrossRef]
- Pichardo, L.A.; Contreras, I.J.; Zamudio, S.R.; Mixcoha, E.; Mendoza, J.G. Synaptic Vesicle Protein 2A as a novel pharmacological target with broad potential for new antiepileptic drugs. In Antiepelieptic Drug Discovery: Novel Approaches; Talevi, A., Rocha, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 53–65. [Google Scholar]
- Klitgaard, H.; Matagne, A.; Grimee, R.; Vanneste-Goemaere, J.; Margineanu, D.G. Electrophysiological, neurochemical and regional effects of levetiracetam in the rat pilocarpine model of temporal lobe epilepsy. Seizure 2003, 12, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Margineanu, D.G.; Matagne, A.; Kaminski, R.M.; Klitgaard, H. Effects of chronic treatment with levetiracetam on hippocampal field responses after pilocarpine-induced status epilepticus in rats. Brain Res. Bull. 2008, 77, 282–285. [Google Scholar] [CrossRef]
- Queiroz, C.M.; Gorter, J.A.; Lopes da Silva, F.H.; Wadman, W.J. Dynamics of evoked local field potentials in the hippocampus of epileptic rats with spontaneous seizures. J. Neurophysiol. 2009, 101, 1588–1597. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glien, M.; Brandt, C.; Potschka, H.; Loscher, W. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia 2002, 43, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Pichardo Macias, L.A.; Ramirez Mendiola, B.A.; Contreras Garcia, I.J.; Zamudio Hernandez, S.R.; Chavez Pacheco, J.L.; Sanchez Huerta, K.B.; Mendoza Torreblanca, J.G. Effect of levetiracetam on extracellular amino acid levels in the dorsal hippocampus of rats with temporal lobe epilepsy. Epilepsy Res. 2018, 140, 111–119. [Google Scholar] [CrossRef]
- Zamudio, S.R.; Pichardo-Macias, L.A.; Diaz-Villegas, V.; Flores-Navarrete, I.L.; Guzman-Velazquez, S. Subchronic cerebrolysin treatment alleviates cognitive impairments and dendritic arborization alterations of granular neurons in the hippocampal dentate gyrus of rats with temporal lobe epilepsy. Epilepsy Behav. 2019, 97, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Arida, R.M.; Scorza, F.A.; Peres, C.A.; Cavalheiro, E.A. The course of untreated seizures in the pilocarpine model of epilepsy. Epilepsy Res. 1999, 34, 99–107. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates Sixth Edition, 6th ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Sanchez-Huerta, K.; Pacheco-Rosado, J.; Gilbert, M.E. Adult onset-hypothyroidism: Alterations in hippocampal field potentials in the dentate gyrus are largely associated with anaesthesia-induced hypothermia. J. Neuroendocrinol. 2015, 27, 8–19. [Google Scholar] [CrossRef]
- Aidil-Carvalho, M.F.; Carmo, A.J.S.; Ribeiro, J.A.; Cunha-Reis, D. Mismatch novelty exploration training enhances hippocampal synaptic plasticity: A tool for cognitive stimulation? Neurobiol. Learn. Mem. 2017, 145, 240–250. [Google Scholar] [CrossRef]
- Kimura, F.; Itami, C.; Ikezoe, K.; Tamura, H.; Fujita, I.; Yanagawa, Y.; Obata, K.; Ohshima, M. Fast activation of feedforward inhibitory neurons from thalamic input and its relevance to the regulation of spike sequences in the barrel cortex. J. Physiol. 2010, 588, 2769–2787. [Google Scholar] [CrossRef]
- Gilbert, M.E.; Burdette, L.J. Hippocampal field potentials: A model system to characterize neurologist. In Neurotoxicology: Approaches and Methods; Chang, L.S.W., Ed.; Academic Press: New York, NY, USA, 1995; pp. 183–202. [Google Scholar]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Loscher, W. Valproate: A reappraisal of its pharmacodynamic properties and mechanisms of action. Prog. Neurobiol. 1999, 58, 31–59. [Google Scholar] [CrossRef]
- Ji-qun, C.; Ishihara, K.; Nagayama, T.; Serikawa, T.; Sasa, M. Long-lasting antiepileptic effects of levetiracetam against epileptic seizures in the spontaneously epileptic rat (SER): Differentiation of levetiracetam from conventional antiepileptic drugs. Epilepsia 2005, 46, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Daoudal, G.; Debanne, D. Long-term plasticity of intrinsic excitability: Learning rules and mechanisms. Learn. Mem. 2003, 10, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, B.D.; Sloviter, R.S. Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats: Implications for hippocampal epileptogenesis. J. Comp. Neurol. 2005, 488, 442–463. [Google Scholar] [CrossRef] [PubMed]
- Cavarsan, C.F.; Malheiros, J.; Hamani, C.; Najm, I.; Covolan, L. Is Mossy Fiber Sprouting a Potential Therapeutic Target for Epilepsy? Front. Neurol. 2018, 9, 1023. [Google Scholar] [CrossRef] [Green Version]
- De Lanerolle, N.C.; Kim, J.H.; Robbins, R.J.; Spencer, D.D. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989, 495, 387–395. [Google Scholar] [CrossRef]
- Magloczky, Z.; Wittner, L.; Borhegyi, Z.; Halasz, P.; Vajda, J.; Czirjak, S.; Freund, T.F. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience 2000, 96, 7–25. [Google Scholar] [CrossRef]
- Meehan, A.L.; Yang, X.; Yuan, L.L.; Rothman, S.M. Levetiracetam has an activity-dependent effect on inhibitory transmission. Epilepsia 2012, 53, 469–476. [Google Scholar] [CrossRef]
- Ohno, Y.; Okumura, T.; Terada, R.; Ishihara, S.; Serikawa, T.; Sasa, M. Kindling-associated SV2A expression in hilar GABAergic interneurons of the mouse dentate gyrus. Neurosci. Lett. 2012, 510, 93–98. [Google Scholar] [CrossRef]
- Tokudome, K.; Okumura, T.; Shimizu, S.; Mashimo, T.; Takizawa, A.; Serikawa, T.; Terada, R.; Ishihara, S.; Kunisawa, N.; Sasa, M.; et al. Synaptic vesicle glycoprotein 2A (SV2A) regulates kindling epileptogenesis via GABAergic neurotransmission. Sci. Rep. 2016, 6, 27420. [Google Scholar] [CrossRef]
- Ohno, Y.; Tokudome, K. Therapeutic Role of Synaptic Vesicle Glycoprotein 2A (SV2A) in Modulating Epileptogenesis. CNS Neurol. Disord. Drug Targets 2017, 16, 463–471. [Google Scholar] [CrossRef]
- Ohno, Y.; Ishihara, S.; Terada, R.; Kikuta, M.; Sofue, N.; Kawai, Y.; Serikawa, T.; Sasa, M. Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem. Biophys. Res. Commun. 2009, 390, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Bronzino, J.D.; Blaise, J.H.; Morgane, P.J. The paired-pulse index: A measure of hippocampal dentate granule cell modulation. Ann. Biomed. Eng. 1997, 25, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Goussakov, I.V.; Fink, K.; Elger, C.E.; Beck, H. Metaplasticity of mossy fiber synaptic transmission involves altered release probability. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 3434–3441. [Google Scholar] [CrossRef]
- Soukupova, M.; Binaschi, A.; Falcicchia, C.; Zucchini, S.; Roncon, P.; Palma, E.; Magri, E.; Grandi, E.; Simonato, M. Impairment of GABA release in the hippocampus at the time of the first spontaneous seizure in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2014, 257, 39–49. [Google Scholar] [CrossRef]
- Zhan, R.Z.; Nadler, J.V. Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J. Neurophysiol. 2009, 102, 670–681. [Google Scholar] [CrossRef] [Green Version]
- Magloczky, Z.; Freund, T.F. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005, 28, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Blitz, D.M.; Foster, K.A.; Regehr, W.G. Short-term synaptic plasticity: A comparison of two synapses. Nat. Rev. Neurosci. 2004, 5, 630–640. [Google Scholar] [CrossRef]
- Upreti, C.; Otero, R.; Partida, C.; Skinner, F.; Thakker, R.; Pacheco, L.F.; Zhou, Z.Y.; Maglakelidze, G.; Veliskova, J.; Velisek, L.; et al. Altered neurotransmitter release, vesicle recycling and presynaptic structure in the pilocarpine model of temporal lobe epilepsy. Brain J. Neurol. 2012, 135, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Soukupova, M.; Binaschi, A.; Falcicchia, C.; Palma, E.; Roncon, P.; Zucchini, S.; Simonato, M. Increased extracellular levels of glutamate in the hippocampus of chronically epileptic rats. Neuroscience 2015, 301, 246–253. [Google Scholar] [CrossRef]
- Gorter, J.A.; Van Vliet, E.A.; Proper, E.A.; De Graan, P.N.; Ghijsen, W.E.; Lopes Da Silva, F.H.; Aronica, E. Glutamate transporters alterations in the reorganizing dentate gyrus are associated with progressive seizure activity in chronic epileptic rats. J. Comp. Neurol. 2002, 442, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Zubareva, O.E.; Kovalenko, A.A.; Kalemenev, S.V.; Schwarz, A.P.; Karyakin, V.B.; Zaitsev, A.V. Alterations in mRNA expression of glutamate receptor subunits and excitatory amino acid transporters following pilocarpine-induced seizures in rats. Neurosci. Lett. 2018, 686, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Nusser, Z.; Mody, I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 2002, 87, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42 (Suppl. 3), 8–12. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.C.; Williams, J.M. Properties and mechanisms of LTP maintenance. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2003, 9, 463–474. [Google Scholar] [CrossRef]
- Cavalheiro, E.A.; Fernandes, M.J.; Turski, L.; Naffah-Mazzacoratti, M.G. Spontaneous recurrent seizures in rats: Amino acid and monoamine determination in the hippocampus. Epilepsia 1994, 35, 1–11. [Google Scholar] [CrossRef]
- Egbenya, D.L.; Hussain, S.; Lai, Y.C.; Xia, J.; Anderson, A.E.; Davanger, S. Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol. Cell. Neurosci. 2018, 92, 93–103. [Google Scholar] [CrossRef]
- Tang, F.R.; Lee, W.L.; Gao, H.; Chen, Y.; Loh, Y.T.; Chia, S.C. Expression of different isoforms of protein kinase C in the rat hippocampus after pilocarpine-induced status epilepticus with special reference to CA1 area and the dentate gyrus. Hippocampus 2004, 14, 87–98. [Google Scholar] [CrossRef]
- Liu, J.X.; Liu, Y.; Tang, F.R. Pilocarpine-induced status epilepticus alters hippocampal PKC expression in mice. Acta Neurobiol. Exp. 2011, 71, 220–232. [Google Scholar]
- Saito, N.; Shirai, Y. Protein kinase C gamma (PKC gamma): Function of neuron specific isotype. J. Biochem. 2002, 132, 683–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Huang, W.; Chen, S.; Lin, M.; Huang, Q.; Huang, H. The Role of 5-HTR6 in Mossy Fiber Sprouting: Activating Fyn and p-ERK1/2 in Pilocarpine-Induced Chronic Epileptic Rats. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.G.; O’Dell, T.J.; Karl, K.A.; Stein, P.L.; Soriano, P.; Kandel, E.R. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 1992, 258, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Pozdnyakova, N.; Dudarenko, M.; Borisova, T. Age-Dependency of Levetiracetam Effects on Exocytotic GABA Release from Nerve Terminals in the Hippocampus and Cortex in Norm and After Perinatal Hypoxia. Cell. Mol. Neurobiol. 2019, 39, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Allone, C.; Lo Buono, V.; Corallo, F.; Pisani, L.R.; Pollicino, P.; Bramanti, P.; Marino, S. Neuroimaging and cognitive functions in temporal lobe epilepsy: A review of the literature. J. Neurol. Sci. 2017, 381, 7–15. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, C.L.; Liu, H.G.; Wang, X.; Zhang, X.; Meng, F.G.; Zhang, J.G. Abnormal hippocampal functional network and related memory impairment in pilocarpine-treated rats. Epilepsia 2018, 59, 1785–1795. [Google Scholar] [CrossRef] [Green Version]
- Smolensky, I.V.; Zubareva, O.E.; Kalemenev, S.V.; Lavrentyeva, V.V.; Dyomina, A.V.; Karepanov, A.A.; Zaitsev, A.V. Impairments in cognitive functions and emotional and social behaviors in a rat lithium-pilocarpine model of temporal lobe epilepsy. Behav. Brain Res. 2019, 372, 112044. [Google Scholar] [CrossRef]
- Operto, F.F.; Pastorino, G.M.G.; Mazza, R.; Roccella, M.; Carotenuto, M.; Margari, L.; Verrotti, A. Cognitive profile in BECTS treated with levetiracetam: A 2-year follow-up. Epilepsy Behav. 2019, 97, 187–191. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-H, G.; Contreras-García, I.J.; Sánchez-Huerta, K.; Queiroz, C.M.T.; Gallardo Gudiño, L.R.; Mendoza-Torreblanca, J.G.; Zamudio, S.R. Levetiracetam Reduced the Basal Excitability of the Dentate Gyrus without Restoring Impaired Synaptic Plasticity in Rats with Temporal Lobe Epilepsy. Brain Sci. 2020, 10, 634. https://doi.org/10.3390/brainsci10090634
González-H G, Contreras-García IJ, Sánchez-Huerta K, Queiroz CMT, Gallardo Gudiño LR, Mendoza-Torreblanca JG, Zamudio SR. Levetiracetam Reduced the Basal Excitability of the Dentate Gyrus without Restoring Impaired Synaptic Plasticity in Rats with Temporal Lobe Epilepsy. Brain Sciences. 2020; 10(9):634. https://doi.org/10.3390/brainsci10090634
Chicago/Turabian StyleGonzález-H, Guillermo, Itzel Jatziri Contreras-García, Karla Sánchez-Huerta, Claudio M. T. Queiroz, Luis Ricardo Gallardo Gudiño, Julieta G. Mendoza-Torreblanca, and Sergio R. Zamudio. 2020. "Levetiracetam Reduced the Basal Excitability of the Dentate Gyrus without Restoring Impaired Synaptic Plasticity in Rats with Temporal Lobe Epilepsy" Brain Sciences 10, no. 9: 634. https://doi.org/10.3390/brainsci10090634
APA StyleGonzález-H, G., Contreras-García, I. J., Sánchez-Huerta, K., Queiroz, C. M. T., Gallardo Gudiño, L. R., Mendoza-Torreblanca, J. G., & Zamudio, S. R. (2020). Levetiracetam Reduced the Basal Excitability of the Dentate Gyrus without Restoring Impaired Synaptic Plasticity in Rats with Temporal Lobe Epilepsy. Brain Sciences, 10(9), 634. https://doi.org/10.3390/brainsci10090634