
Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons


 , , ,
, , ,  ,
, 
{kind=link}
Abstract
:1. Introduction
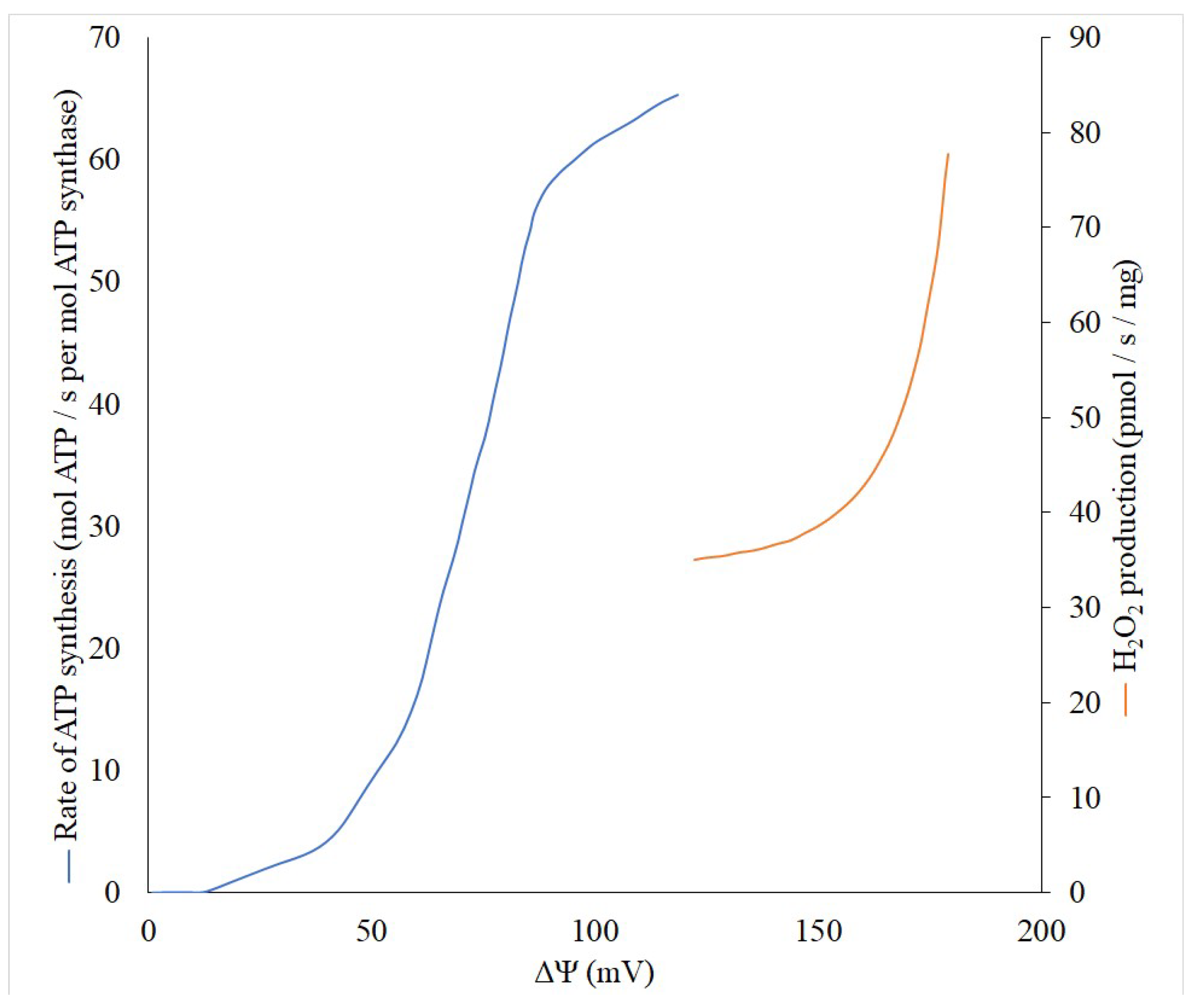
2. Pros
2.1. Anti-Oxygen
2.2. Anti-ROS
2.3. Anti-Obesity
2.4. Increased CO2 Production
2.5. Increased Mitophagic Activity
2.6. Increased Mitochondrial Biogenesis
2.7. Enhanced AMPK Signaling
2.8. Anti-Inflammatory
3. Cons
3.1. Possibility of Local Hypoxia/Ischemia
3.2. The Drop of ATP Synthesis
3.3. Acidic Shift
3.4. The Drop of the Membrane Potential-Dependent Reactions
3.5. Diminished Mitophagic Activity?
3.6. Hyperactivation of Oxidative Metabolism, Loss of Cellular Reserves
3.7. Reduction of Redox Signaling
3.8. Thermogenesis
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Fernanda, M.C.; Caldeira da Silva, C.C.; Fernanda, M.C.; Alicia, J.K. Mild Mitochondrial Uncoupling as a Therapeutic Strategy. Curr. Drug Targets 2011, 12, 783–789. [Google Scholar]
- Liu, D.; Zhang, Y.; Gharavi, R.; Park, H.R.; Lee, J.; Siddiqui, S.; Telljohann, R.; Nassar, M.R.; Cutler, R.G.; Becker, K.G.; et al. The Mitochondrial Uncoupler DNP Triggers Brain Cell MTOR Signaling Network Reprogramming and CREB Pathway Up-Regulation. J. Neurochem. 2015, 134, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Dorighello, G.G.; Rovani, J.C.; Paim, B.A.; Rentz, T.; Assis, L.H.P.; Vercesi, A.E.; Oliveira, H.C.F. Mild Mitochondrial Uncoupling Decreases Experimental Atherosclerosis, A Proof of Concept. J. Atheroscler. Thromb. 2021. [Google Scholar] [CrossRef]
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild Mitochondrial Uncoupling Protects from Ionizing Radiation Induced Cell Death by Attenuating Oxidative Stress and Mitochondrial Damage. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148325. [Google Scholar] [CrossRef]
- Berkowitz, B.A.; Olds, H.K.; Richards, C.; Joy, J.; Rosales, T.; Podolsky, R.H.; Childers, K.L.; Hubbard, W.B.; Sullivan, P.G.; Gao, S.; et al. Novel Imaging Biomarkers for Mapping the Impact of Mild Mitochondrial Uncoupling in the Outer Retina in Vivo. PLoS ONE 2020, 15, e0226840. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childress, E.S.; Alexopoulos, S.J.; Hoehn, K.L.; Santos, W.L. Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential. J. Med. Chem. 2018, 61, 4641–4655. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Jankauskas, S.S.; Rokitskaya, T.I.; Chupyrkina, A.A.; Pevzner, I.B.; Zorova, L.D.; Isaev, N.K.; Antonenko, Y.N.; Skulachev, V.P.; et al. Mild Uncoupling of Respiration and Phosphorylation as a Mechanism Providing Nephro- and Neuroprotective Effects of Penetrating Cations of the SkQ Family. Biochem. Mosc. 2012, 77, 1029–1037. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Denisov, S.S.; Silachev, D.N.; Khailova, L.S.; Jankauskas, S.S.; Rokitskaya, T.I.; Danilina, T.I.; Kotova, E.A.; Korshunova, G.A.; Plotnikov, E.Y.; et al. A Long-Linker Conjugate of Fluorescein and Triphenylphosphonium as Mitochondria-Targeted Uncoupler and Fluorescent Neuro- and Nephroprotector. Biochim. Biophys. Acta 2016, 1860, 2463–2473. [Google Scholar] [CrossRef]
- Silachev, D.N.; Khailova, L.S.; Babenko, V.A.; Gulyaev, M.V.; Kovalchuk, S.I.; Zorova, L.D.; Plotnikov, E.Y.; Antonenko, Y.N.; Zorov, D.B. Neuroprotective Effect of Glutamate-Substituted Analog of Gramicidin a Is Mediated by the Uncoupling of Mitochondria. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 3434–3442. [Google Scholar] [CrossRef]
- Ost, M.; Keipert, S.; Klaus, S. Targeted Mitochondrial Uncoupling beyond UCP1—The Fine Line between Death and Metabolic Health. Biochimie 2017, 134, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of Coupling of Oxidative Phosphorylation and the Membrane Potential of Mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Uncoupling: New Approaches to an Old Problem of Bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124. [Google Scholar] [CrossRef] [Green Version]
- Caldeira da Silva, C.C.; Cerqueira, F.M.; Barbosa, L.F.; Medeiros, M.H.G.; Kowaltowski, A.J. Mild Mitochondrial Uncoupling in Mice Affects Energy Metabolism, Redox Balance and Longevity. Aging Cell 2008, 7, 552–560. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.G.; Dilger, J.P. Transport of Protons across Membranes by Weak Acids. Physiol. Rev. 1980, 60, 825–863. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Volkov, N.I. Carboxyatractylate Inhibits the Uncoupling Effect of Free Fatty Acids. FEBS Lett. 1988, 226, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Samartsev, V.N.; Smirnov, A.V.; Zeldi, I.P.; Markova, O.V.; Mokhova, E.N.; Skulachev, V.P. Involvement of Aspartate/Glutamate Antiporter in Fatty Acid-Induced Uncoupling of Liver Mitochondria. Biochim. Biophys. Acta (BBA) Bioenerg. 1997, 1319, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Firsov, A.M.; Popova, L.B.; Khailova, L.S.; Nazarov, P.A.; Kotova, E.A.; Antonenko, Y.N. Protonophoric Action of BAM15 on Planar Bilayers, Liposomes, Mitochondria, Bacteria and Neurons. Bioelectrochemistry 2021, 137, 107673. [Google Scholar] [CrossRef] [PubMed]
- Khailova, L.S.; Vygodina, T.V.; Lomakina, G.Y.; Kotova, E.A.; Antonenko, Y.N. Bicarbonate Suppresses Mitochondrial Membrane Depolarization Induced by Conventional Uncouplers. Biochem. Biophys. Res. Commun. 2020, 530, 29–34. [Google Scholar] [CrossRef]
- Starkov, A.A.; Dedukhova, V.I.; Skulachev, V.P. 6-Ketocholestanol Abolishes the Effect of the Most Potent Uncouplers of Oxidative Phosphorylation in Mitochondria. FEBS Lett. 1994, 355, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Rousset, S.; Alves-Guerra, M.-C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.-M.; Bouillaud, F.; Ricquier, D. The Biology of Mitochondrial Uncoupling Proteins. Diabetes 2004, 53, S130–S135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, R.M. Oxygen: The Poison Is in the Dose. Transfusion 2013, 53, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The Rise of Oxygen in Earth’s Early Ocean and Atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef]
- Jones, D.P. Intracellular Diffusion Gradients of O2 and ATP. Am. J. Physiol. Cell Physiol. 1986, 250, C663–C675. [Google Scholar] [CrossRef]
- Näpänkangas, J.P.; Liimatta, E.V.; Joensuu, P.; Bergmann, U.; Ylitalo, K.; Hassinen, I.E. Superoxide Production during Ischemia-Reperfusion in the Perfused Rat Heart: A Comparison of Two Methods of Measurement. J. Mol. Cell Cardiol. 2012, 53, 906–915. [Google Scholar] [CrossRef]
- Waypa, G.B.; Schumacker, P.T. O2 Sensing in Hypoxic Pulmonary Vasoconstriction: The Mitochondrial Door Re-Opens. Respir. Physiol. Neurobiol. 2002, 132, 81–91. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A.; Fiskum, G. Regulation of Brain Mitochondrial H2O2 Production by Membrane Potential and NAD(P)H Redox State. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef]
- Skulachev, V.P. Anion Carriers in Fatty Acid-Mediated Physiological Uncoupling. J. Bioenerg. Biomembr. 1999, 31, 431–445. [Google Scholar] [CrossRef]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating Cation/Fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668. [Google Scholar] [CrossRef] [Green Version]
- Trendeleva, T.A.; Sukhanova, E.I.; Rogov, A.G.; Zvyagilskaya, R.A.; Seveina, I.I.; Ilyasova, T.M.; Cherepanov, D.A.; Skulachev, V.P. Role of Charge Screening and Delocalization for Lipophilic Cation Permeability of Model and Mitochondrial Membranes. Mitochondrion 2013, 13, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Kaim, G.; Dimroth, P. ATP Synthesis by F-Type ATP Synthase Is Obligatorily Dependent on the Transmembrane Voltage. EMBO J. 1999, 18, 4118–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrauwen, P.; Walder, K.; Ravussin, E. Human Uncoupling Proteins and Obesity. Obes. Res. 1999, 7, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-Y.; Beretta, M.; Alexopoulos, S.J.; Shah, D.P.; Olzomer, E.M.; Hargett, S.R.; Childress, E.S.; Salamoun, J.M.; Aleksovska, I.; Roseblade, A.; et al. Mitochondrial Uncoupler SHC517 Reverses Obesity in Mice without Affecting Food Intake. Metabolism 2021, 117, 154724. [Google Scholar] [CrossRef]
- Axelrod, C.L.; King, W.T.; Davuluri, G.; Noland, R.C.; Hall, J.; Hull, M.; Dantas, W.S.; Zunica, E.R.; Alexopoulos, S.J.; Hoehn, K.L.; et al. BAM15-Mediated Mitochondrial Uncoupling Protects against Obesity and Improves Glycemic Control. EMBO Mol. Med. 2020, 12, e12088. [Google Scholar] [CrossRef] [PubMed]
- Tainter, M.L.; Stockton, A.B.; Cutting, W.C. Dinitrophenol in the treatment of obesity: Final report. J. Am. Med. Assoc. 1935, 105, 332–337. [Google Scholar] [CrossRef]
- Ruiz-Ramírez, A.; López-Acosta, O.; Barrios-Maya, M.A.; El-Hafidi, M. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid. Med. Cell. Longev. 2016, 2016, e9340654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasho, V.N.; Boyer, P.D. Relationships of Inosine Triphosphate and Bicarbonate Effects on F1 ATPase to the Binding Change Mechanism. J. Bioenerg. Biomembr. 1984, 16, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A Common Target for Genetic Mutations and Environmental Toxicants in Parkinson’s Disease. Front. Genet. 2017, 8, 177. [Google Scholar] [CrossRef]
- Kumar, S.; Flacke, J.-P.; Kostin, S.; Appukuttan, A.; Reusch, H.P.; Ladilov, Y. SLC4A7 Sodium Bicarbonate Co-Transporter Controls Mitochondrial Apoptosis in Ischaemic Coronary Endothelial Cells. Cardiovasc. Res. 2011, 89, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Alka, K.; Casey, J.R. Bicarbonate Transport in Health and Disease. IUBMB Life 2014, 66, 596–615. [Google Scholar] [CrossRef]
- Nozik-Grayck, E.; Huang, Y.-C.T.; Carraway, M.S.; Piantadosi, C.A. Bicarbonate-Dependent Superoxide Release and Pulmonary Artery Tone. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2327–H2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohzuki, M.; Tomimatsu, T.; Fukuda, H.; Kanagawa, T.; Kanzaki, T.; Shimoya, K.; Murata, Y. Long-Term Neuroprotective Effects of Carbon Dioxide on Neonatal Rat Hypoxic-Ischemic Brain Injury: An Experimental Study of Skilled Motor Tasks. Am. J. Obstet. Gynecol. 2006, 195, 240–245. [Google Scholar] [CrossRef]
- Deng, R.-M.; Liu, Y.-C.; Li, J.-Q.; Xu, J.-G.; Chen, G. The Role of Carbon Dioxide in Acute Brain Injury. Med. Gas. Res. 2020, 10, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Azad, P.; Zhao, H.W.; Wang, J.; Poulsen, O.; Freitas, B.C.; Muotri, A.R.; Haddad, G.G. The Na+/HCO3- Co-Transporter Is Protective during Ischemia in Astrocytes. Neuroscience 2016, 339, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Shu, Y.; Wang, J.; Brinkman, B.C.; Haddad, G.G. Factors Influencing Cell Fate in the Infarct Rim. J. Neurochem. 2007, 100, 1224–1233. [Google Scholar] [CrossRef]
- Sohn, Y.; Yoo, K.-Y.; Park, O.K.; Kwon, S.-H.; Lee, C.H.; Choi, J.H.; Hwang, I.K.; Seo, J.Y.; Cho, J.H.; Won, M.-H. Na+/HCO3- Cotransporter Immunoreactivity Changes in Neurons and Expresses in Astrocytes in the Gerbil Hippocampal CA1 Region after Ischemia/Reperfusion. Neurochem. Res. 2011, 36, 2459–2469. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.S.; Paris, A.; Codron, P.; Cassereau, J.; Procaccio, V.; Lenaers, G.; Reynier, P.; Chevrollier, A. Current Mechanistic Insights into the CCCP-Induced Cell Survival Response. Biochem. Pharmacol. 2018, 148, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 View of ANT Function in Mitochondrial Biology and Necrotic Cell Death. J. Mol. Cell Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ye, M.; Liu, D.; Yang, J.; Ding, J.-W.; Zhang, J.; Wang, X.-A.; Dong, W.-S.; Fan, Z.-X.; Yang, J. UCP2 Protect the Heart from Myocardial Ischemia/Reperfusion Injury via Induction of Mitochondrial Autophagy. J. Cell Biochem. 2019, 120, 15455–15466. [Google Scholar] [CrossRef]
- Schlagowski, A.I.; Singh, F.; Charles, A.L.; Gali Ramamoorthy, T.; Favret, F.; Piquard, F.; Geny, B.; Zoll, J. Mitochondrial Uncoupling Reduces Exercise Capacity despite Several Skeletal Muscle Metabolic Adaptations. J. Appl. Physiol. 2014, 116, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itami, N.; Shiratsuki, S.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Mitochondrial Biogenesis and Degradation Are Induced by CCCP Treatment of Porcine Oocytes. Reproduction 2015, 150, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- De Pauw, A.; Demine, S.; Tejerina, S.; Dieu, M.; Delaive, E.; Kel, A.; Renard, P.; Raes, M.; Arnould, T. Mild Mitochondrial Uncoupling Does Not Affect Mitochondrial Biogenesis but Downregulates Pyruvate Carboxylase in Adipocytes: Role for Triglyceride Content Reduction. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1123–E1141. [Google Scholar] [CrossRef] [Green Version]
- Kansaku, K.; Takeo, S.; Itami, N.; Kin, A.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Maternal Aging Affects Oocyte Resilience to Carbonyl Cyanide-m-Chlorophenylhydrazone -Induced Mitochondrial Dysfunction in Cows. PLoS ONE 2017, 12, e0188099. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Keeping the Home Fires Burning: AMP-Activated Protein Kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Keipert, S.; Rossmeisl, M.; Kopecky, J. Augmenting Energy Expenditure by Mitochondrial Uncoupling: A Role of AMP-Activated Protein Kinase. Genes Nutr. 2012, 7, 369–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Li, T.; Ji, T.; Yi, W.; Yang, Z.; Wang, S.; Yang, Y.; Gu, C. AMPK: Potential Therapeutic Target for Ischemic Stroke. Theranostics 2018, 8, 4535–4551. [Google Scholar] [CrossRef]
- McCullough, L.D.; Zeng, Z.; Li, H.; Landree, L.E.; McFadden, J.; Ronnett, G.V. Pharmacological Inhibition of AMP-Activated Protein Kinase Provides Neuroprotection in Stroke *. J. Biol. Chem. 2005, 280, 20493–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, Z.; Viollet, B.; Ronnett, G.V.; McCullough, L.D. Neuroprotective Effects of Adenosine Monophosphate- Activated Protein Kinase Inhibition and Gene Deletion in Stroke. Stroke 2007, 38, 2992–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manwani, B.; McCullough, L.D. Function of the Master Energy Regulator Adenosine Monophosphate-Activated Protein Kinase in Stroke. J. Neurosci. Res. 2013, 91, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.P.; Issara-Amphorn, J.; Charoensappakit, A.; Udompornpitak, K.; Bhunyakarnjanarat, T.; Saisorn, W.; Sae-Khow, K.; Leelahavanichkul, A. BAM15, a Mitochondrial Uncoupling Agent, Attenuates Inflammation in the LPS Injection Mouse Model: An Adjunctive Anti-Inflammation on Macrophages and Hepatocytes. J. Innate Immun. 2021, 1–17. [Google Scholar] [CrossRef]
- Patoli, D.; Mignotte, F.; Deckert, V.; Dusuel, A.; Dumont, A.; Rieu, A.; Jalil, A.; Dongen, K.V.; Bourgeois, T.; Gautier, T.; et al. Inhibition of Mitophagy Drives Macrophage Activation and Antibacterial Defense during Sepsis. J. Clin. Investig. 2020, 130, 5858–5874. [Google Scholar] [CrossRef]
- Busceti, C.L.; Cotugno, M.; Bianchi, F.; Forte, M.; Stanzione, R.; Marchitti, S.; Battaglia, G.; Nicoletti, F.; Fornai, F.; Rubattu, S. Brain Overexpression of Uncoupling Protein-2 (UCP2) Delays Renal Damage and Stroke Occurrence in Stroke-Prone Spontaneously Hypertensive Rats. Int. J. Mol. Sci. 2020, 21, 4289. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-L.; Xu, F.-Y.; Ji, J.-J.; Song, P.; Pei, Y.-Q.; He, M.-J.; Wang, Z.-C.; You, S.-J.; Hua, Z.-C.; Cheng, J.; et al. Activation of UCP2 by Anethole Trithione Suppresses Neuroinflammation after Intracerebral Hemorrhage. Acta Pharm. Sin. 2021. [Google Scholar] [CrossRef]
- Pan, X.; Song, Y.; He, M.; Yan, X.; Huang, C.; Li, J.; Dong, W.; Cheng, J.; Jia, J. Mitochondrial Uncouplers Confer Protection by Activating AMP-Activated Protein Kinase to Inhibit Neuroinflammation Following Intracerebral Hemorrhage. Biol. Pharm. Bull. 2020, 43, 1210–1219. [Google Scholar] [CrossRef]
- Kety, S.S.; Schmidt, C.F. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J. Clin. Investig. 1948, 27, 484–492. [Google Scholar] [CrossRef]
- Krogh, A. The Supply of Oxygen to the Tissues and the Regulation of the Capillary Circulation. J. Physiol. 1919, 52, 457–474. [Google Scholar] [CrossRef]
- Yaniv, Y.; Juhaszova, M.; Nuss, H.B.; Wang, S.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Matching ATP Supply and Demand in Mammalian Heart. Ann. N. Y. Acad. Sci. 2010, 1188, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Burmester, T.; Hankeln, T. Function and Evolution of Vertebrate Globins. Acta Physiol. 2014, 211, 501–514. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Wittenberg, B.A. Myoglobin-Enhanced Oxygen Delivery to Isolated Cardiac Mitochondria. J. Exp. Biol. 2007, 210, 2082–2090. [Google Scholar] [CrossRef] [Green Version]
- Burmester, T.; Hankeln, T. What Is the Function of Neuroglobin? J. Exp. Biol. 2009, 212, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cohen, J.; Boron, W.F.; Schulten, K.; Tajkhorshid, E. Exploring Gas Permeability of Cellular Membranes and Membrane Channels with Molecular Dynamics. J. Struct. Biol. 2007, 157, 534–544. [Google Scholar] [CrossRef]
- Xie, L.-K.; Yang, S.-H. Brain Globins in Physiology and Pathology. Med. Gas. Res. 2016, 6, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A.; Sylvia, A.L.; Jöbsis, F.F. Cyanide-Induced Cytochrome a,A3 Oxidation-Reduction Responses in Rat Brain in Vivo. J. Clin. Investig. 1983, 72, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaresima, V.; Springett, R.; Cope, M.; Wyatt, J.T.; Delpy, D.T.; Ferrari, M.; Cooper, C.E. Oxidation and Reduction of Cytochrome Oxidase in the Neonatal Brain Observed by in Vivo Near-Infrared Spectroscopy. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1366, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Kreisman, N.R.; Sick, T.J.; Lamanna, J.C.; Rosenthal, M. Local Tissue Oxygen Tension—Cytochrome a,A3 Redox Relationships in Rat Cerebral Cortex in Vivo. Brain Res. 1981, 218, 161–174. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. Biol. Rev. 1966, 41, 445–501. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, Y. Energy Conservation and Uncoupling in Mitochondria. J. Supramol. Struct. 1975, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. Mechanisms of Energy Transformations. Annu. Rev. Biochem. 1977, 46, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L.; Eska, T.; Morris, H.P.; Catterall, W.A. Deficiency of Uncoupler-Stimulated Adenosine Triphosphatase Activity in Tightly Coupled Hepatoma Mitochondria. Proc. Natl. Acad. Sci. USA 1971, 68, 1079–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silachev, D.N.; Gulyaev, M.V.; Zorova, L.D.; Khailova, L.S.; Gubsky, L.V.; Pirogov, Y.A.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Magnetic Resonance Spectroscopy of the Ischemic Brain under Lithium Treatment. Link to Mitochondrial Disorders under Stroke. Chem. Biol. Interact. 2015, 237, 175–182. [Google Scholar] [CrossRef]
- Rizack, M.A. Activation of an epinephrine-sensitive lipolytic activity from adipose tissue by adenosine 3′,5′-phosphate. J. Biol. Chem. 1964, 239, 392–395. [Google Scholar] [CrossRef]
- Eastman, A. Deoxyribonuclease II in Apoptosis and the Significance of Intracellular Acidification. Cell Death Differ. 1994, 1, 7–9. [Google Scholar] [PubMed]
- Zorov, D.B.; Andrianova, N.V.; Babenko, V.A.; Bakeeva, L.E.; Zorov, S.D.; Zorova, L.D.; Pevsner, I.B.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N. Nonphosphorylating Oxidation in Mitochondria and Related Processes. Biochemistry 2020, 85, 1570–1577. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Hay, R.; Böhni, P.; Gasser, S. How Mitochondria Import Proteins. Biochim. Biophys. Acta 1984, 779, 65–87. [Google Scholar] [CrossRef]
- Pfanner, N.; Tropschug, M.; Neupert, W. Mitochondrial Protein Import: Nucleoside Triphosphates Are Involved in Conferring Import-Competence to Precursors. Cell 1987, 49, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Neupert, W. Protein Import into Mitochondria. Annu. Rev. Biochem. 1997, 66, 863–917. [Google Scholar] [CrossRef]
- Truscott, K.N.; Pfanner, N.; Voos, W. Transport of proteins into mitochondria. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2001; pp. 81–136. ISBN 978-3-540-44510-4. [Google Scholar]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial Membrane Potential Regulates PINK1 Import and Proteolytic Destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Popkov, V.A.; Zorova, L.D.; Vorobjev, I.A.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Jankauskas, S.S.; Babenko, V.A.; Plotnikov, E.Y. Mitochondrial Aging: Is There a Mitochondrial Clock? J. Gerontol. Biol. Sci. Med. Sci. 2017, 72, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunter, K.K.; Gunter, T.E. Transport of Calcium by Mitochondria. J. Bioenerg. Biomembr. 1994, 26, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by Which Mitochondria Transport Calcium. Am. J. Physiol. Cell Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, Redox Environment and ROS Emission in Heart Failure: Two Sides of the Same Coin? J. Mol. Cell. Cardiol. 2021, 151, 113–125. [Google Scholar] [CrossRef]
- Maragos, W.F.; Korde, A.S. Mitochondrial Uncoupling as a Potential Therapeutic Target in Acute Central Nervous System Injury. J. Neurochem. 2004, 91, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-Induced Neuron Death Requires Mitochondrial Calcium Uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Khodorov, B.I.; Storozhevykh, T.P.; Surin, A.M.; Yuryavichyus, A.I.; Sorokina, E.G.; Borodin, A.V.; Vinskaya, N.P.; Khaspekov, L.G.; Pinelis, V.G. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci. Behav. Physiol. 2002, 32, 541–547. [Google Scholar] [CrossRef]
- Korde, A.S.; Sullivan, P.G.; Maragos, W.F. The Uncoupling Agent 2,4-Dinitrophenol Improves Mitochondrial Homeostasis Following Striatal Quinolinic Acid Injections. J. Neurotrauma 2005, 22, 1142–1149. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and Aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Silachev, D.N.; Andrianova, N.V.; Pevzner, I.B.; Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Zorov, D.B. Aged Kidney: Can We Protect It? Autophagy, Mitochondria and Mechanisms of Ischemic Preconditioning. Cell Cycle 2018, 17, 1291–1309. [Google Scholar] [CrossRef] [PubMed]
- Berezhnov, A.V.; Soutar, M.P.M.; Fedotova, E.I.; Frolova, M.S.; Plun-Favreau, H.; Zinchenko, V.P.; Abramov, A.Y. Intracellular Ph modulates autophagy and mitophagy. J. Biol. Chem. 2016, 291, 8701–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Fielden, L.F.; Stojanovski, D. Mitochondrial Protein Transport in Health and Disease. Semin. Cell Dev. Biol. 2018, 76, 142–153. [Google Scholar] [CrossRef]
- Attardi, G.; Schatz, G. Biogenesis of Mitochondria. Annu. Rev. Cell Biol. 1988, 4, 289–333. [Google Scholar] [CrossRef]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 2019, 73, 1028.e5–1043.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Transport of Proteins into Mitochondria: A Potassium Diffusion Potential Is Able to Drive the Import of ADP/ATP Carrier. EMBO J. 1985, 4, 2819–2825. [CrossRef]
- Martin, J.; Mahlke, K.; Pfanner, N. Role of an Energized Inner Membrane in Mitochondrial Protein Import. Delta Psi Drives the Movement of Presequences. J. Biol. Chem. 1991, 266, 18051–18057. [Google Scholar] [CrossRef]
- Geissler, A.; Krimmer, T.; Bömer, U.; Guiard, B.; Rassow, J.; Pfanner, N. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b(2) modulates the deltapsi-dependence of translocation of the matrix-targeting sequence. Mol. Biol. Cell 2000, 11, 3977–3991. [Google Scholar] [CrossRef]
- Bauer, M.F.; Sirrenberg, C.; Neupert, W.; Brunner, M. Role of Tim23 as Voltage Sensor and Presequence Receptor in Protein Import into Mitochondria. Cell 1996, 87, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Maley, G.F.; Lardy, H.A. Metabolic Effects of Thyroid Hormones in Vitro. II. Influence of Thyroxine and Triiodothyronine on Oxidative Phosphorylation. J. Biol. Chem. 1953, 204, 435–444. [Google Scholar] [CrossRef]
- Zorov, D.B.; Bannikova, S.Y.; Belousov, V.V.; Vyssokikh, M.Y.; Zorova, L.D.; Isaev, N.K.; Krasnikov, B.F.; Plotnikov, E.Y. Reactive Oxygen and Nitrogen Species: Friends or Foes? Biochemistry 2005, 70, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Babenko, V.A.; Zorov, S.D.; Popkov, V.A.; Jankauskas, S.S.; Zinchenko, V.P.; Sukhikh, G.T.; et al. The Mitochondrion as a Key Regulator of Ischaemic Tolerance and Injury. HeartLung Circ. 2014, 23, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Chien, L.-F.; Ainscow, E.K.; Rolfe, D.F.S.; Porter, R.K. The Causes and Functions of Mitochondrial Proton Leak. Biochim. Biophys. Acta (BBA) Bioenerg. 1994, 1187, 132–139. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Maslov, S.P.; Sivkova, V.G.; Kalinichenko, L.P.; Maslova, G.M. Cold uncoupling of oxidative phosphorylation in the muscles of white mice. Biokhimiia 1963, 28, 70–79. [Google Scholar]
- Nedergaard, J.; Cannon, B. Chapter 9—Brown adipose tissue as a heat-producing thermoeffector. In Handbook of Clinical Neurology; Thermoregulation: From Basic Neuroscience to Clinical Neurology Part I; Romanovsky, A.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 156, pp. 137–152. [Google Scholar] [CrossRef]
- Himms-Hagen, J. Cellular Thermogenesis. Annu. Rev. Physiol. 1976, 38, 315–351. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.E. Thermogenic Mechanisms and Their Hormonal Regulation. Physiol. Rev. 2006, 86, 435–464. [Google Scholar] [CrossRef]
- Pressman, B.C.; Lardy, H.A. Effect of Surface Active Agents on the Latent ATPASE of Mitochondira. Biochim. Biophys. Acta 1956, 21, 458–466. [Google Scholar] [CrossRef]
- Skulachev, V.P. Fatty Acid Circuit as a Physiological Mechanism of Uncoupling of Oxidative Phosphorylation. FEBS Lett. 1991, 294, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P.; Sharaf, A.A.; Liberman, E.A. Proton Conductors in the Respirator Chain and Artificial Membranes. Nature 1967, 216, 718–719. [Google Scholar] [CrossRef] [PubMed]
- Jeẑek, P.; Garlid, K.D. Mammalian Mitochondrial Uncoupling Proteins. Int. J. Biochem. Cell Biol. 1998, 30, 1163–1168. [Google Scholar] [CrossRef]
- Samartsev, V.N.; Simonyan, R.A.; Markova, O.V.; Mokhova, E.N.; Skulachev, V.P. Comparative Study on Uncoupling Effects of Laurate and Lauryl Sulfate on Rat Liver and Skeletal Muscle Mitochondria. Biochim. Biophys. Acta (BBA) Bioenerg. 2000, 1459, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Tsofina, L.M.; Volkov, N.I.; Vygodina, T.V. The ATP/ADP-Antiporter Is Involved in the Uncoupling Effect of Fatty Acids on Mitochondria. Eur. J. Biochem. 1989, 182, 585–592. [Google Scholar] [CrossRef]
- White, M.G.; Luca, L.E.; Nonner, D.; Saleh, O.; Hu, B.; Barrett, E.F.; Barrett, J.N. Cellular Mechanisms of Neuronal Damage From Hyperthermia. Prog. Brain Res. 2007, 162, 347–371. [Google Scholar] [CrossRef]
- Simon, H.B. Hyperthermia. N. Engl. J. Med. 1993, 329, 483–487. [Google Scholar] [CrossRef]
- Walter, E.J.; Carraretto, M. The Neurological and Cognitive Consequences of Hyperthermia. Crit. Care 2016, 20, 199. [Google Scholar] [CrossRef] [Green Version]
- Kopec, K.T.; Kim, T.; Mowry, J.; Aks, S.; Kao, L. Role of Dantrolene in Dinitrophenol (DNP) Overdose: A Continuing Question? Am. J. Emerg. Med. 2019, 37, 1216.e1–1216.e2. [Google Scholar] [CrossRef]
- Du, F.; Zhu, L.; Qian, Z.-M.; Wu, X.-M.; Yung, W.-H.; Ke, Y. Hyperthermic Preconditioning Protects Astrocytes from Ischemia/Reperfusion Injury by up-Regulation of HIF-1 Alpha Expression and Binding Activity. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Aibiki, M.; Nagoya, J. Neuroprotective Effects of Hyperthermic Preconditioning on Infarcted Volume after Middle Cerebral Artery Occlusion in Rats: Role of Adenosine Receptors. Crit. Care Med. 2002, 30, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Lin, M.-T. Heat Shock Protein Expression Protects Against Cerebral Ischemia and Monoamine Overload in Rat Heatstroke. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H1961–H1967. [Google Scholar] [CrossRef]
- Sun, Y.-J.; Zhang, Z.-Y.; Fan, B.; Li, G.-Y. Neuroprotection by Therapeutic Hypothermia. Front. Neurosci. 2019, 13, 586. [Google Scholar] [CrossRef]
- Pamenter, M.E.; Lau, G.Y.; Richards, J.G. Effects of Cold on Murine Brain Mitochondrial Function. PLoS ONE 2018, 13, e0208453. [Google Scholar] [CrossRef]
- Skulachev, V.P. Role of Uncoupled and Non-Coupled Oxidations in Maintenance of Safely Low Levels of Oxygen and Its One-Electron Reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.G. 2,4 Dinitrophenol as Medicine. Cells 2019, 8, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G.; Wasilewska-Sampaio, A.P.; Barbosa, A.C.A.P.; Gomes, F.C.A.; Ferreira, S.T. Cyclic AMP Enhancers and Aβ Oligomerization Blockers as Potential Therapeutic Agents in Alzheimers Disease. Curr. Alzheimer Res. 2007, 4, 263–271. [Google Scholar]
- Sebollela, A.; Freitas-Corrêa, L.; Oliveira, F.F.; Mendes, C.T.; Wasilewska-Sampaio, A.P.; Camacho-Pereira, J.; Galina, A.; Brentani, H.; Passetti, F.; De Felice, F.G.; et al. Expression Profile of Rat Hippocampal Neurons Treated with the Neuroprotective Compound 2,4-Dinitrophenol: Up-Regulation of CAMP Signaling Genes. Neurotox. Res. 2010, 18, 112–123. [Google Scholar] [CrossRef]
- Gohel, D.; Singh, R. Mitohormesis; Potential Implications in Neurodegenerative Diseases. Mitochondrion 2021, 56, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tapia, P.C. Sublethal Mitochondrial Stress with an Attendant Stoichiometric Augmentation of Reactive Oxygen Species May Precipitate Many of the Beneficial Alterations in Cellular Physiology Produced by Caloric Restriction, Intermittent Fasting, Exercise and Dietary Phytonutrients: “Mitohormesis” for Health and Vitality. Med. Hypotheses 2006, 66, 832–843. [Google Scholar] [CrossRef]
- Murry, C.E.; Richard, V.J.; Jennings, R.B.; Reimer, K.A. Myocardial Protection Is Lost Before Contractile Function Recovers from Ischemic Preconditioning. Am. J. Physiol. Heart Circ. Physiol. 1991, 260, H796–H804. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kato, H.; Nakata, N.; Kogure, K. Protection of Rat Hippocampus against Ischemic Neuronal Damage by Pretreatment with Sublethal Ischemia. Brain Res. 1992, 586, 121–124. [Google Scholar] [CrossRef]
- Pashkovskaya, A.A.; Vazdar, M.; Zimmermann, L.; Jovanovic, O.; Pohl, P.; Pohl, E.E. Mechanism of Long-Chain Free Fatty Acid Protonation at the Membrane-Water Interface. Biophys. J. 2018, 114, 2142–2151. [Google Scholar] [CrossRef] [Green Version]
- Jezek, P. Fatty Acid Interaction with Mitochondrial Uncoupling Proteins. J. Bioenerg. Biomembr. 1999, 31, 457–466. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Wojtczak, L. Involvement of the Dicarboxylate Carrier in the Protonophoric Action of Long-Chain Fatty Acids in Mitochondria. Biochem. Biophys. Res. Commun. 1997, 232, 414–417. [Google Scholar] [CrossRef]
- Hodges, J.M.; Gutenstein, M.; Marx, W. Thyroxine and Yeast Metabolism: Uncoupling of Phosphorylation. Arch. Biochem. Biophys. 1963, 101, 429–435. [Google Scholar] [CrossRef]
- Klemperer, H.G. The Uncoupling of Oxidative Phosphorylation in Rat-Liver Mitochondria by Thyroxine, Triiodothyronine and Related Substances. Biochem. J. 1955, 60, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, F.L.; Lipmann, F. The Uncoupling of Respiration and Phosphorylation by Thyroid Hormones. Proc. Natl. Acad. Sci. USA 1954, 40, 909–921. [Google Scholar] [CrossRef] [Green Version]
- Yau, W.W.; Singh, B.K.; Lesmana, R.; Zhou, J.; Sinha, R.A.; Wong, K.A.; Wu, Y.; Bay, B.-H.; Sugii, S.; Sun, L.; et al. Thyroid Hormone (T3) Stimulates Brown Adipose Tissue Activation via Mitochondrial Biogenesis and MTOR-Mediated Mitophagy. Autophagy 2019, 15, 131–150. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast Bioenergetics to Classify Amyotrophic Lateral Sclerosis Patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors Correlated with Hypermetabolism in Patients with Amyotrophic Lateral Sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Sarnat, H.B.; Flores-Sarnat, L.; Hader, W.; Bello-Espinosa, L. Mitochondrial “Hypermetabolic” Neurons in Paediatric Epileptic Foci. Can. J. Neurol. Sci 2011, 38, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.; Barja, G. ADP-Regulation of Mitochondrial Free Radical Production Is Different with Complex I- or Complex II-Linked Substrates: Implications for the Exercise Paradox and Brain Hypermetabolism. J. Bioenerg. Biomembr. 1997, 29, 241–249. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, C.T.; Rothwell, N.J.; Shrewsbury-Gee, J. Sympathetically Mediated Hypermetabolic Response to Cerebral Ischemia in the Rat. Can. J. Physiol. Pharm. 1990, 68, 1334–1337. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Nedergaard, J. Mitochondrial (‘mild’) Uncoupling and ROS Production: Physiologically Relevant or Not? Biochem. Soc. Trans. 2011, 39, 1305–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorov, D.B.; Andrianova, N.V.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Zorov, S.D.; Zorova, L.D.; Plotnikov, E.Y.; Sukhikh, G.T.; Silachev, D.N. Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sci. 2021, 11, 1050. https://doi.org/10.3390/brainsci11081050
Zorov DB, Andrianova NV, Babenko VA, Pevzner IB, Popkov VA, Zorov SD, Zorova LD, Plotnikov EY, Sukhikh GT, Silachev DN. Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sciences. 2021; 11(8):1050. https://doi.org/10.3390/brainsci11081050
Chicago/Turabian StyleZorov, Dmitry B., Nadezda V. Andrianova, Valentina A. Babenko, Irina B. Pevzner, Vasily A. Popkov, Savva D. Zorov, Ljubava D. Zorova, Egor Yu. Plotnikov, Gennady T. Sukhikh, and Denis N. Silachev. 2021. "Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons" Brain Sciences 11, no. 8: 1050. https://doi.org/10.3390/brainsci11081050
APA StyleZorov, D. B., Andrianova, N. V., Babenko, V. A., Pevzner, I. B., Popkov, V. A., Zorov, S. D., Zorova, L. D., Plotnikov, E. Y., Sukhikh, G. T., & Silachev, D. N. (2021). Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sciences, 11(8), 1050. https://doi.org/10.3390/brainsci11081050