

Microglia PKM2 Mediates Neuroinflammation and Neuron Loss in Mice Epilepsy through the Astrocyte C3-Neuron C3R Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Stereotaxic AAV Microinjection
2.3. PISE Mouse Model
2.4. Primary Cell Culture, Transfection and Treatment
2.5. Immunofluorescence (IF) Analysis
2.6. Multiplex TSA Staining
2.7. Nissl Staining
2.8. Enzyme Linked Immunosorbent Assay (ELISA)
2.9. Hoechst33342 Staining
2.10. Real-Time PCR
- C3 Forward: CTGAACACAGCCAAAGATCGGA;
- C3 Reverse: CCATGTTCAAGTCCTTATGGTCAG;
- IL-1β Forward: AGCTTCCTTGTGCAAGTGTCTG;
- IL-1β Reverse: ATCTTTTGGGGTCCGTCAACTTCA;
- IL-6 Forward: GTTCCTCTCTGCAAGAGACTTC;
- IL-6 Reverse: AGTCTCCTCTCCGGACTTGT;
- TNF-α Forward: TGTAGCCCACGTCGTAGCA;
- TNF-α Reverse: TGGGTGAGGAGCACGTAGT;
- β-actin Forward: GGACTGTTACTGAGCTGCGTT;
- β-actin Reverse: CGCCTTCACCGTTCCAGTT.
2.11. Western Blot
2.12. CCK8 Assay
2.13. Statistical Analysis
3. Results
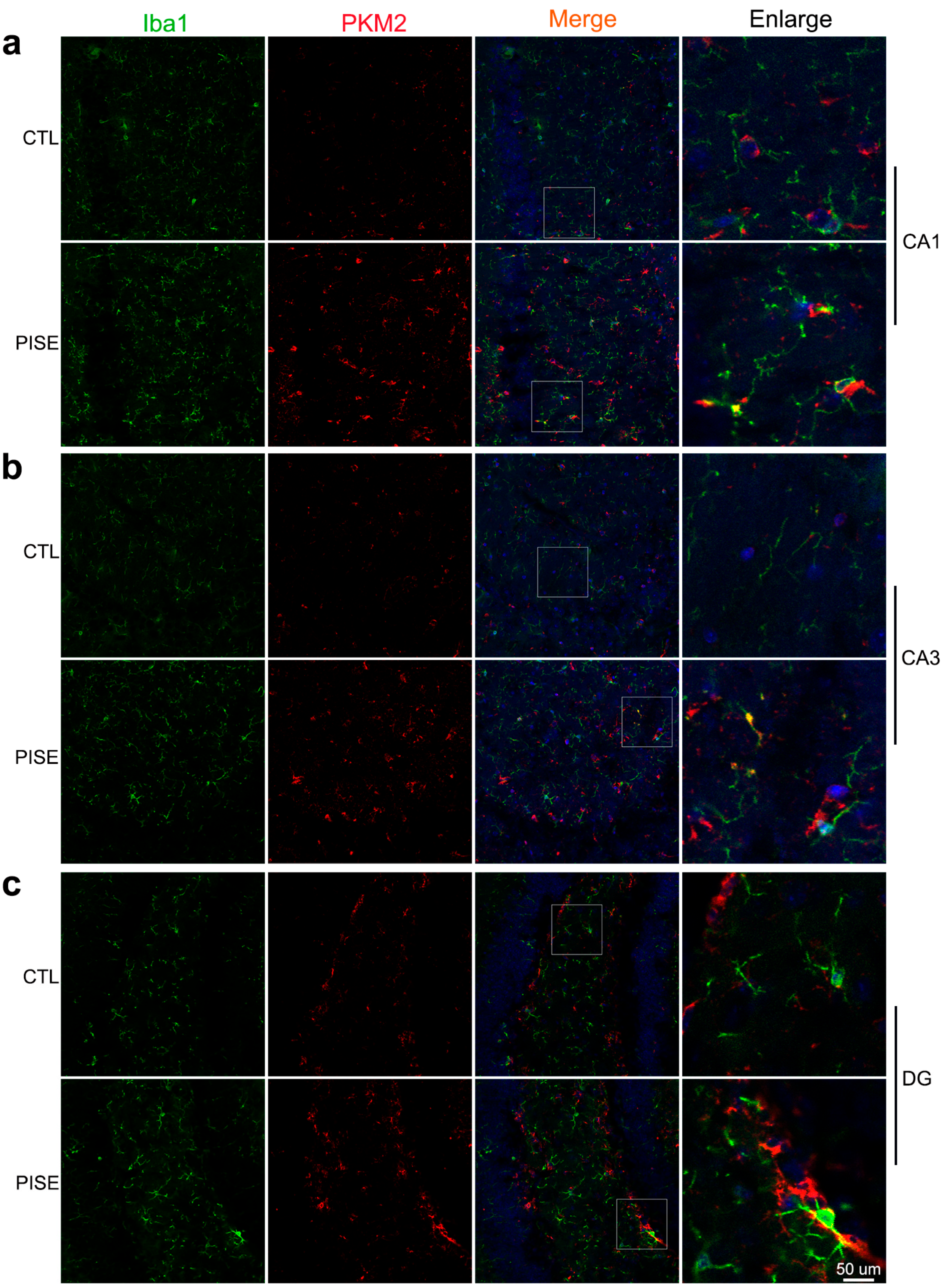
3.1. Microglia PKM2 Is Upregulated in the Hippocampus of PISE Mice Model
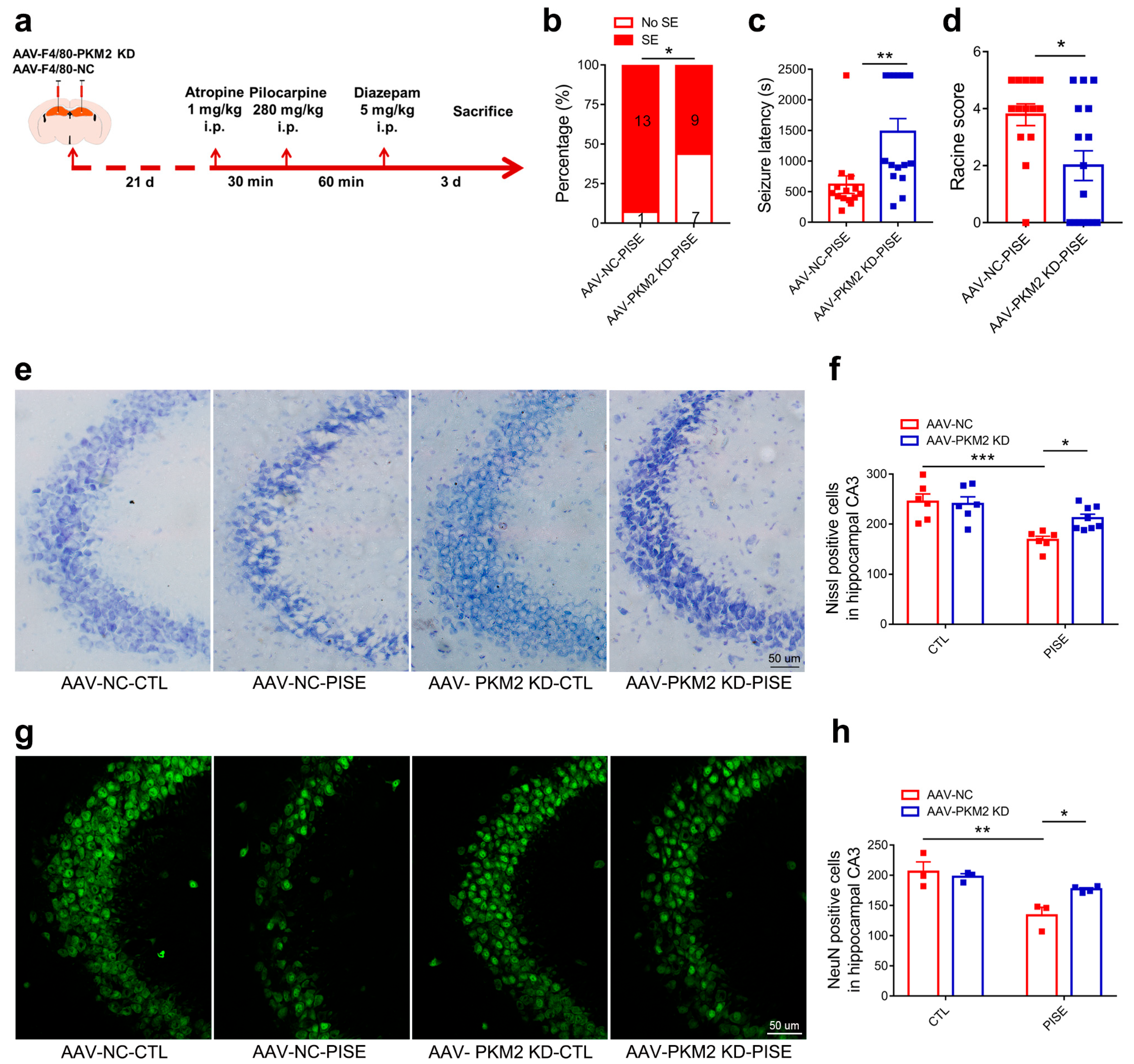
3.2. Microglia PKM2 Knockdown Alleviates PISE Seizure and Rescues Neuron Death in PISE Mice Model
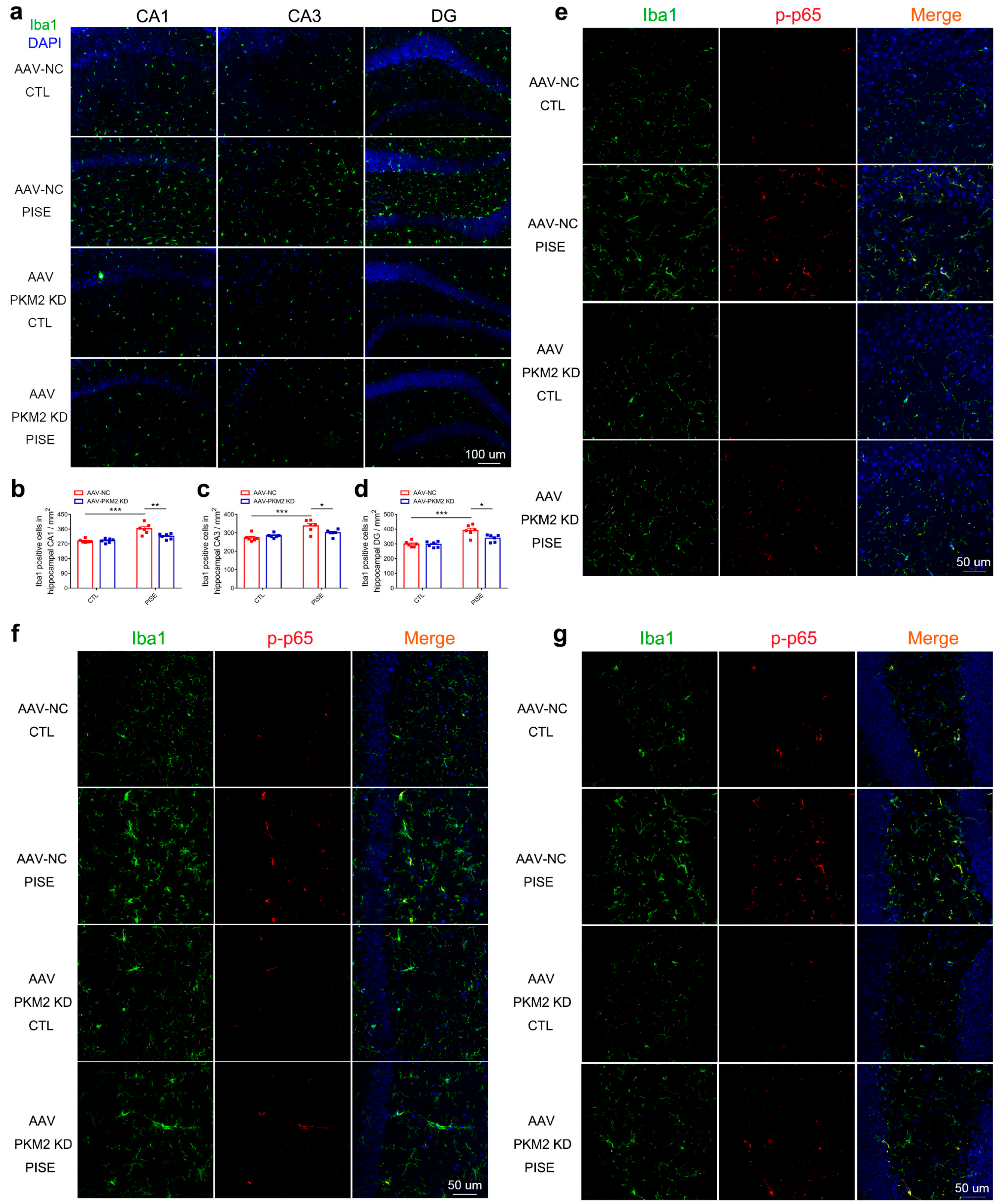
3.3. Microglia PKM2 Knockdown Inhibits Microglia Activation via the Suppression of NF-κB Activation in PISE Mice
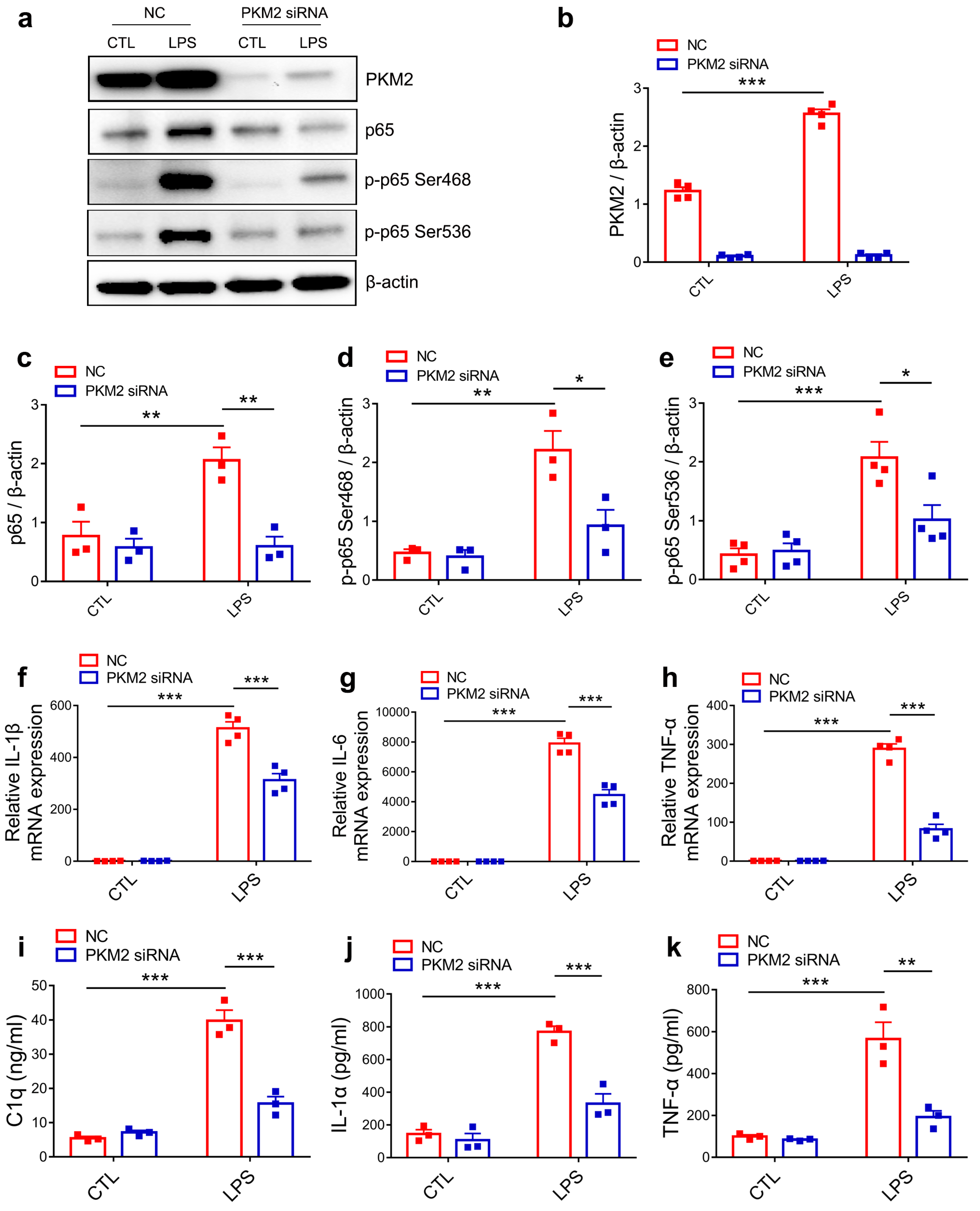
3.4. PKM2 Knockdown Prevents C1q, TNF-α and IL-1α Secretion by Inhibiting NF-κB Activation in LPS-Activated Microglia
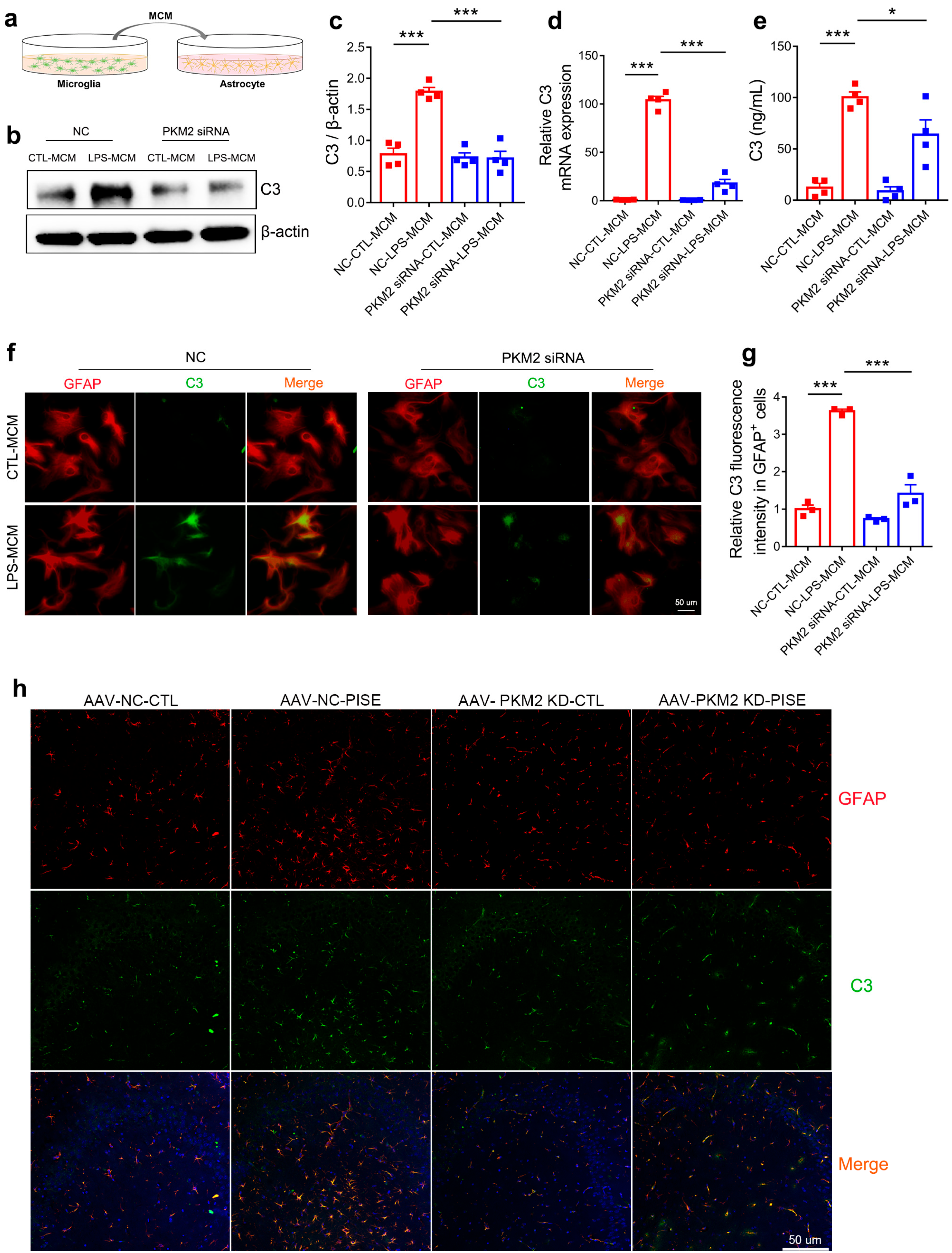
3.5. Microglia PKM2 Knockdown Attenuates Astrocyte C3 Expression
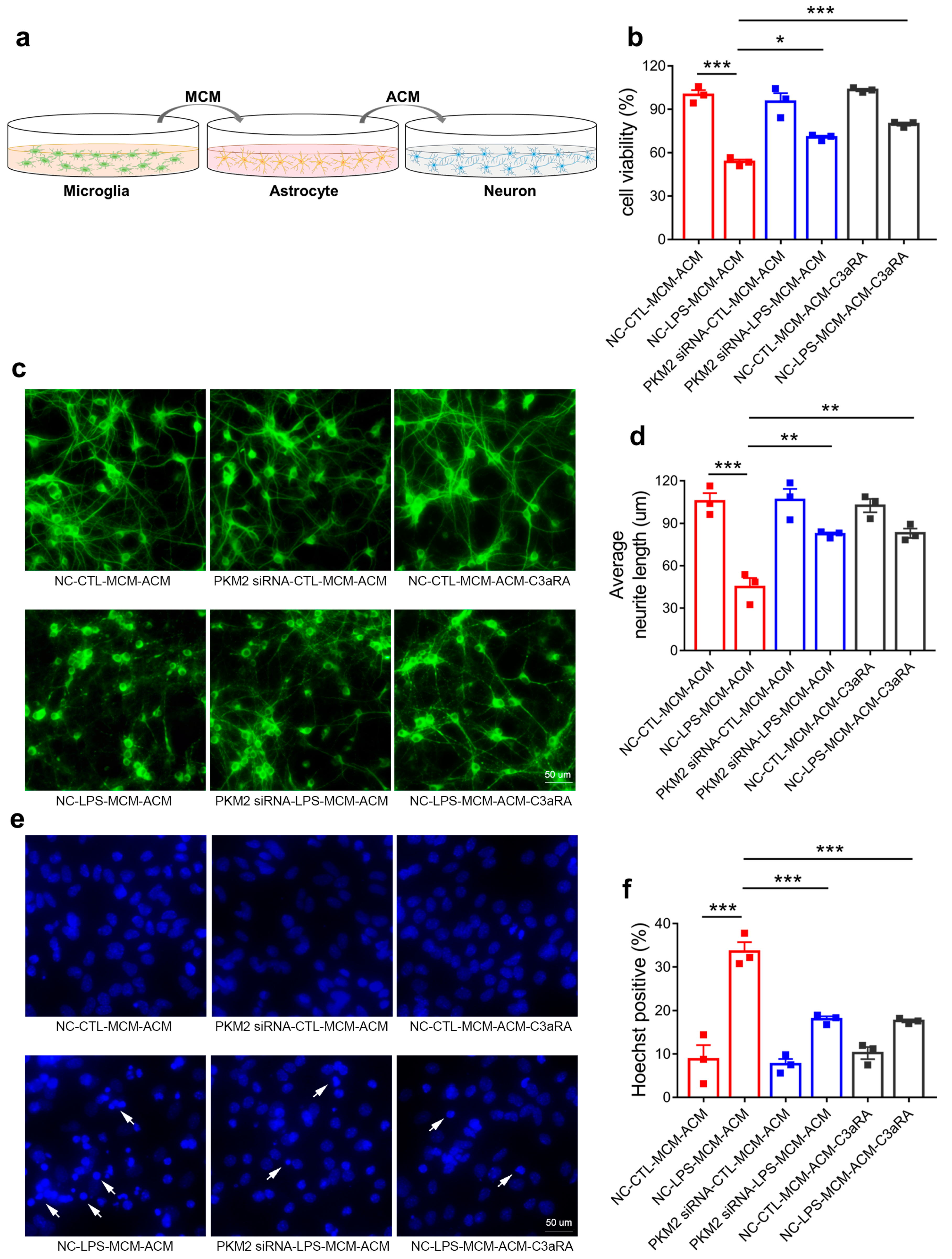
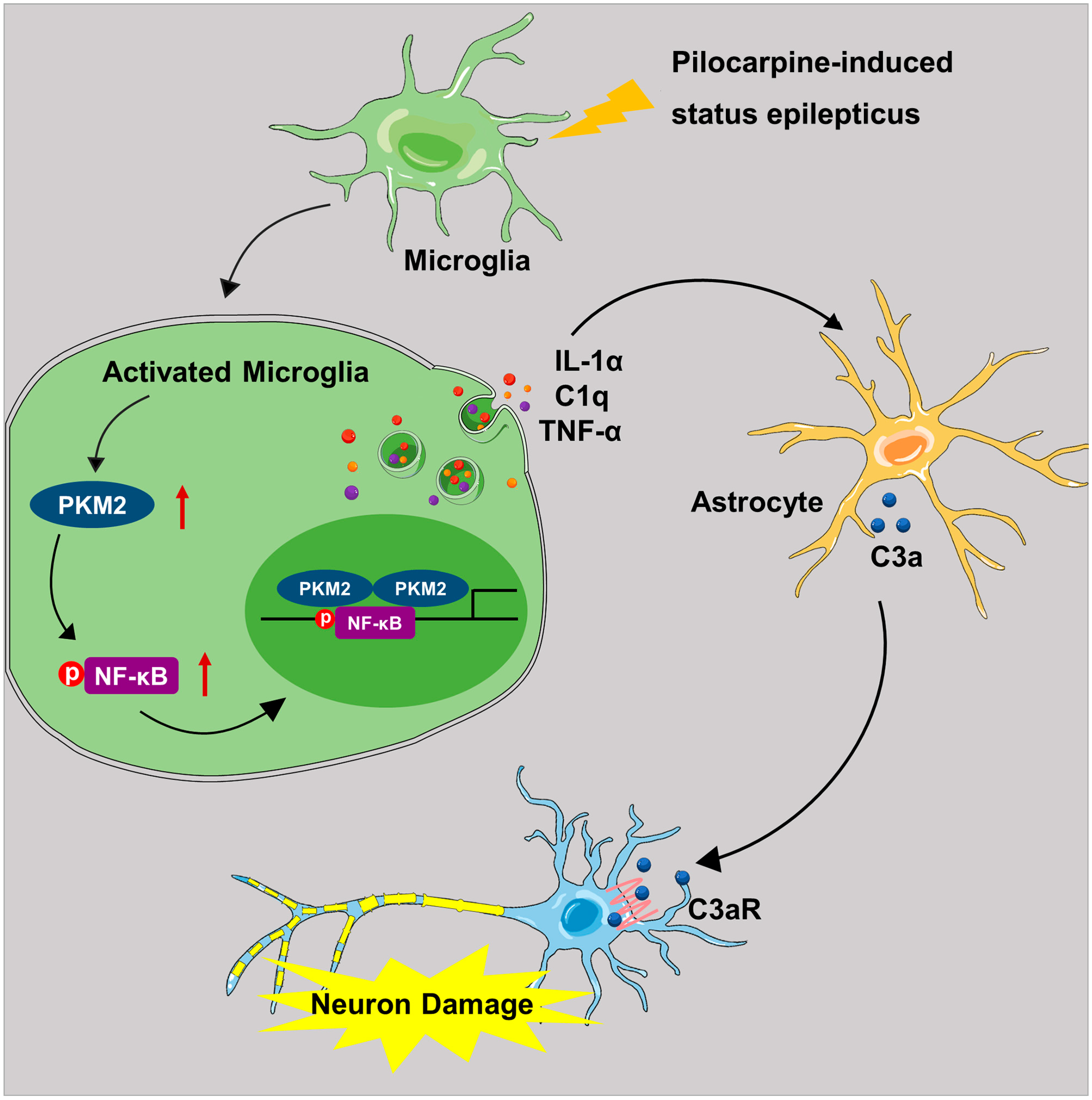
3.6. Microglia PKM2 Knockdown Rescues Neurons via Microglia NF-κB/astrocyte C3/neuron C3aR Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Kuchenbuch, M. Impact of predictive, preventive and precision medicine strategies in epilepsy. Nat. Rev. Neurol. 2020, 16, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharm. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Volnova, A.; Tsytsarev, V.; Ganina, O.; Velez-Crespo, G.E.; Alves, J.M.; Ignashchenkova, A.; Inyushin, M. The Anti-Epileptic Effects of Carbenoxolone In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 663. [Google Scholar] [CrossRef]
- Luan, G.; Gao, Q.; Zhai, F.; Chen, Y.; Li, T. Upregulation of HMGB1, toll-like receptor and RAGE in human Rasmussen’s encephalitis. Epilepsy Res. 2016, 123, 36–49. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Walsh, J.G.; Sinclair, D.B.; Johnson, E.; Tang-Wai, R.; Wheatley, B.M.; Branton, W.; Maingat, F.; Snyder, T.; Gross, D.W.; et al. Inflammasome induction in Rasmussen’s encephalitis: Cortical and associated white matter pathogenesis. J. Neuroinflamm. 2013, 10, 152. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Aronica, E.; Crino, P.B. Inflammation in epilepsy: Clinical observations. Epilepsia 2011, 52 (Suppl. 3), 26–32. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, L.; An, D.; Xu, W.; Wu, C.; Sha, S.; Li, Y.; Zhu, Y.; Chen, A.; Du, Y.; et al. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 2019, 10, 386. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Smith, M.D.; Jin, J.; Garton, T.; Taylor, M.; Chao, A.; Meyers, K.; Kornberg, M.D.; Zack, D.J.; Ohayon, J.; et al. Complement component 3 from astrocytes mediates retinal ganglion cell loss during neuroinflammation. Acta Neuropathol. 2021, 142, 899–915. [Google Scholar] [CrossRef]
- Chen, M.M.; Hu, Z.L.; Ding, J.H.; Du, R.H.; Hu, G. Astrocytic Kir6.1 deletion aggravates neurodegeneration in the lipopolysaccharide-induced mouse model of Parkinson’s disease via astrocyte-neuron cross talk through complement C3-C3R signaling. Brain Behav. Immun. 2021, 95, 310–320. [Google Scholar] [CrossRef]
- Alawieh, A.M.; Langley, E.F.; Feng, W.; Spiotta, A.M.; Tomlinson, S. Complement-Dependent Synaptic Uptake and Cognitive Decline after Stroke and Reperfusion Therapy. J. Neurosci. 2020, 40, 4042–4058. [Google Scholar] [CrossRef]
- Tripathi, A.; Whitehead, C.; Surrao, K.; Pillai, A.; Madeshiya, A.; Li, Y.; Khodadadi, H.; Ahmed, A.O.; Turecki, G.; Baban, B.; et al. Type 1 interferon mediates chronic stress-induced neuroinflammation and behavioral deficits via complement component 3-dependent pathway. Mol. Psychiatry 2021, 26, 3043–3059. [Google Scholar] [CrossRef]
- Dong, X.; Fan, J.; Lin, D.; Wang, X.; Kuang, H.; Gong, L.; Chen, C.; Jiang, J.; Xia, N.; He, D.; et al. Captopril alleviates epilepsy and cognitive impairment by attenuation of C3-mediated inflammation and synaptic phagocytosis. J. Neuroinflamm. 2022, 19, 226. [Google Scholar] [CrossRef]
- Ashizawa, K.; Willingham, M.C.; Liang, C.M.; Cheng, S.Y. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 1991, 266, 16842–16846. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Jin, X.; Zhang, D.; Li, D.; Hao, F.; Feng, Y.; Gu, S.; Meng, F.; Tian, M.; et al. O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc. Natl. Acad. Sci. USA 2017, 114, 13732–13737. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ma, F.; Wang, Y.; Hao, L.; Zeng, H.; Jia, C.; Wang, Y.; Liu, P.; Ong, I.M.; Li, B.; et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017, 19, 1358–1370. [Google Scholar] [CrossRef]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational Modifications of Pyruvate Kinase M2: Tweaks that Benefit Cancer. Front. Oncol. 2018, 8, 22. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Metabolic Kinases Moonlighting as Protein Kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, L.E.A.; Prado, D.S.; Veras, F.P.; Fonseca, M.M.; Toller-Kawahisa, J.E.; Rosa, M.H.; Publio, G.A.; Martins, T.V.; Ramalho, F.S.; Waisman, A.; et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020, 217, e20190613. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, T.; Bosco, D.B.; Xie, M.; Zheng, J.; Dheer, A.; Ying, Y.; Wu, Q.; Lennon, V.A.; Wu, L.J. The complement C3-C3aR pathway mediates microglia-astrocyte interaction following status epilepticus. Glia 2021, 69, 1155–1169. [Google Scholar] [CrossRef]
- Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.M.; Mee, E.W.; Curtis, M.A.; Faull, R.L.; Dragunow, M. M-CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J. Neuroinflamm. 2013, 10, 85. [Google Scholar] [CrossRef]
- Pan, R.Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022, 34, 634–648. [Google Scholar] [CrossRef]
- Li, M.; Lu, H.; Wang, X.; Duan, C.; Zhu, X.; Zhang, Y.; Ge, X.; Ji, F.; Wang, X.; Su, J.; et al. Pyruvate kinase M2 (PKM2) interacts with activating transcription factor 2 (ATF2) to bridge glycolysis and pyroptosis in microglia. Mol. Immunol. 2021, 140, 250–266. [Google Scholar] [CrossRef]
- Lu, L.; Wang, H.; Liu, X.; Tan, L.; Qiao, X.; Ni, J.; Sun, Y.; Liang, J.; Hou, Y.; Dou, H. Pyruvate kinase isoform M2 impairs cognition in systemic lupus erythematosus by promoting microglial synaptic pruning via the beta-catenin signaling pathway. J. Neuroinflamm. 2021, 18, 229. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: From synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef]
- Anders, S.; Minge, D.; Griemsmann, S.; Herde, M.K.; Steinhauser, C.; Henneberger, C. Spatial properties of astrocyte gap junction coupling in the rat hippocampus. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130600. [Google Scholar] [CrossRef]
- Orellana, J.A.; Martinez, A.D.; Retamal, M.A. Gap junction channels and hemichannels in the CNS: Regulation by signaling molecules. Neuropharmacology 2013, 75, 567–582. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Turner, D.A.; Adamson, D.C. Neuronal-astrocyte metabolic interactions: Understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Wang, Y.; He, Y.; Wang, T.; Huang, X.H.; Zhao, C.M.; Zhang, L.; Li, S.W.; Wang, C.; Qu, Y.N.; et al. A1 astrocytes contribute to murine depression-like behavior and cognitive dysfunction, which can be alleviated by IL-10 or fluorocitrate treatment. J. Neuroinflamm. 2020, 17, 200. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Z.; Wang, S.; Ma, L.; Wang, J.; Yu, D.; Zhang, Z.; Wu, L.; Peng, Z.; Liu, W.; et al. Astrocytic A1/A2 paradigm participates in glycogen mobilization mediated neuroprotection on reperfusion injury after ischemic stroke. J. Neuroinflamm. 2021, 18, 230. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.T.; Dawson, V.L.; Dawson, T.M. The A1 astrocyte paradigm: New avenues for pharmacological intervention in neurodegeneration. Mov. Disord. 2019, 34, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Tenner, A.J.; Mollnes, T.E. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharm. Rev. 2021, 73, 792–827. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef]
- Pozo-Rodrigalvarez, A.; Ollaranta, R.; Skoog, J.; Pekny, M.; Pekna, M. Hyperactive Behavior and Altered Brain Morphology in Adult Complement C3a Receptor Deficient Mice. Front. Immunol. 2021, 12, 604812. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353. [Google Scholar] [CrossRef]
- Zheng, J.; Lu, J.; Mei, S.; Wu, H.; Sun, Z.; Fang, Y.; Xu, S.; Wang, X.; Shi, L.; Xu, W.; et al. Ceria nanoparticles ameliorate white matter injury after intracerebral hemorrhage: Microglia-astrocyte involvement in remyelination. J. Neuroinflamm. 2021, 18, 43. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhou, R.; Peng, H.; Peng, J.; Li, Q.; Mei, M. Microglia PKM2 Mediates Neuroinflammation and Neuron Loss in Mice Epilepsy through the Astrocyte C3-Neuron C3R Signaling Pathway. Brain Sci. 2023, 13, 262. https://doi.org/10.3390/brainsci13020262
Li X, Zhou R, Peng H, Peng J, Li Q, Mei M. Microglia PKM2 Mediates Neuroinflammation and Neuron Loss in Mice Epilepsy through the Astrocyte C3-Neuron C3R Signaling Pathway. Brain Sciences. 2023; 13(2):262. https://doi.org/10.3390/brainsci13020262
Chicago/Turabian StyleLi, Xinlin, Rong Zhou, Hui Peng, Jing Peng, Qiaoling Li, and Meng Mei. 2023. "Microglia PKM2 Mediates Neuroinflammation and Neuron Loss in Mice Epilepsy through the Astrocyte C3-Neuron C3R Signaling Pathway" Brain Sciences 13, no. 2: 262. https://doi.org/10.3390/brainsci13020262
APA StyleLi, X., Zhou, R., Peng, H., Peng, J., Li, Q., & Mei, M. (2023). Microglia PKM2 Mediates Neuroinflammation and Neuron Loss in Mice Epilepsy through the Astrocyte C3-Neuron C3R Signaling Pathway. Brain Sciences, 13(2), 262. https://doi.org/10.3390/brainsci13020262