


A Case of Anti-GAD 65 Autoimmune Encephalitis Associated with Focal Segmental Stiff-Person Syndrome

{kind=link}
{kind=link}
Abstract
:1. Introduction
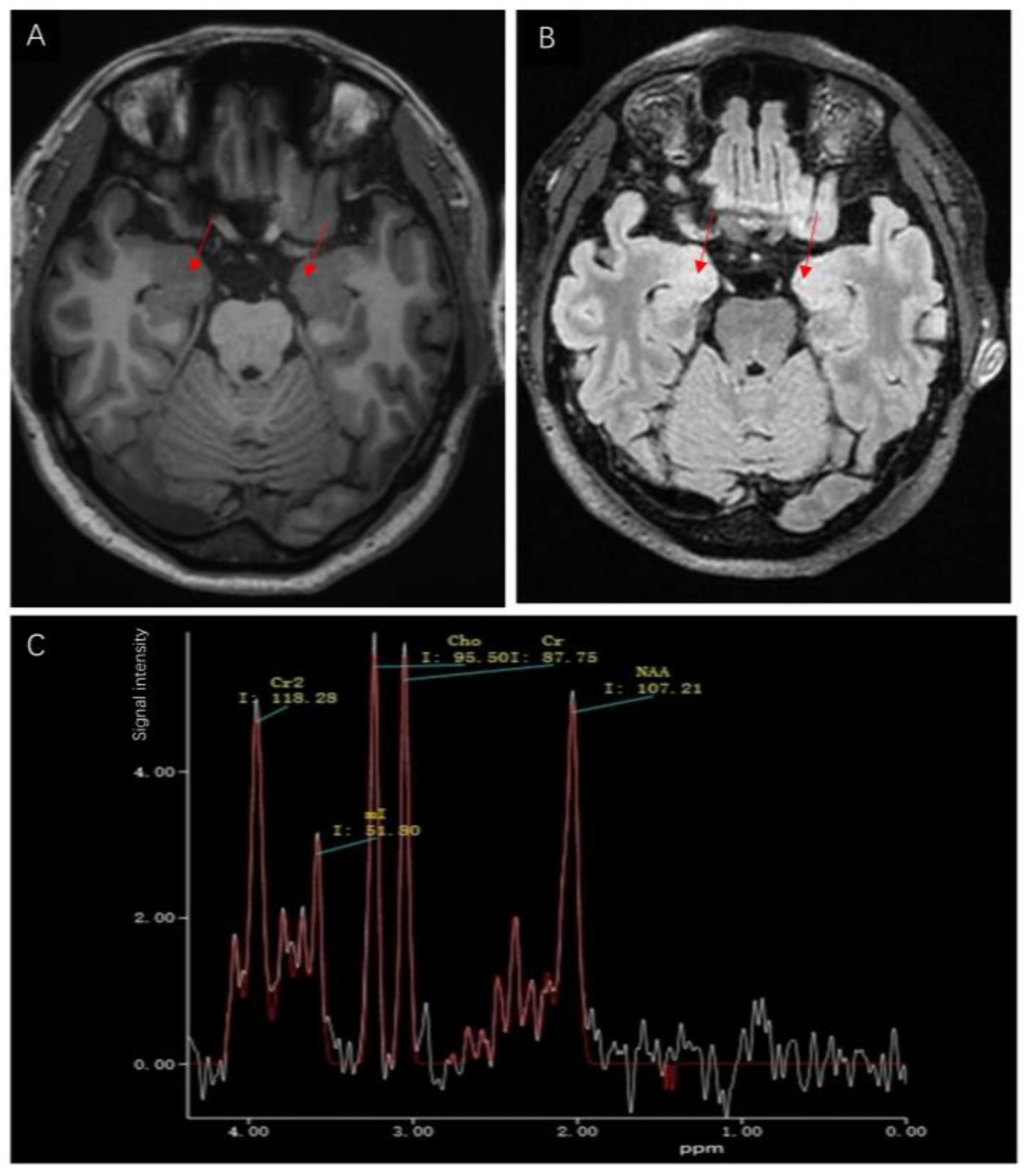
2. Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erlander, M.G.; Tillakaratne, N.J.; Feldblum, S.; Patel, N.; Tobin, A.J. Two genes encode distinct glutamate decarboxylases. Neuron 1991, 7, 91–100. [Google Scholar] [CrossRef]
- Fenalti, G.; Buckle, A.M. Structural biology of the GAD autoantigen. Autoimmun. Rev. 2010, 9, 148–152. [Google Scholar] [CrossRef]
- Liimatainen, S.; Peltola, M.; Sabater, L.; Fallah, M.; Kharazmi, E.; Haapala, A.-M.; Dastidar, P.; Knip, M.; Saiz, A.; Peltola, J. Clinical significance of glutamic acid decarboxylase antibodies in patients with epilepsy. Epilepsia 2010, 51, 760–767. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Dade, M.; Berzero, G.; Izquierdo, C.; Giry, M.; Benazra, M.; Delattre, J.-Y.; Psimaras, D.; Alentorn, A. Neurological Syndromes Associated with Anti-GAD Antibodies. Int. J. Mol. Sci. 2020, 21, 3701. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F. The neurological syndromes associated with glutamic acid decarboxylase antibodies. J. Autoimmun. 2019, 101, 35–47. [Google Scholar] [CrossRef]
- McKeon, A. Stiff-Man Syndrome and Variants. Arch. Neurol. 2012, 69, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Walikonis, J.E.; Lennon, V.A. Radioimmunoassay for Glutamic Acid Decarboxylase (GAD65) Autoantibodies as a Diagnostic Aid for Stiff-Man Syndrome and a Correlate of Susceptibility to Type 1 Diabetes Mellitus. Mayo Clin. Proc. 1998, 73, 1161–1166. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Jankovic, J. Stiff-person syndrome: Insights into a complex autoimmune disorder. J. Neurol. Neurosurg. Psychiatry 2014, 86, 840–848. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Fujii, M.; Li, M.; McElroy, B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology 2000, 55, 1531–1535. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C. Stiff person syndrome: Advances in pathogenesis and therapeutic interventions. Curr. Treat. Options Neurol. 2009, 11, 102–110. [Google Scholar] [CrossRef]
- Dalmau, J.; Tüzün, E.; Wu, H.-Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Shan, W.; Lv, R.; Li, Z.; Wang, Q. Clinical characteristics of GAD 65-associated autoimmune encephalitis. Acta Neurol. Scand. 2020, 142, 281–293. [Google Scholar] [CrossRef]
- Steriade, C.; Britton, J.; Dale, R.C.; Gadoth, A.; Irani, S.R.; Linnoila, J.; McKeon, A.; Shao, X.; Venegas, V.; Bien, C.G. Acute symptomatic seizures secondary to autoimmune encephalitis and autoimmune-associated epilepsy: Conceptual definitions. Epilepsia 2020, 61, 1341–1351. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.J.; Sharpe, L.; Hunt, C.; Gandy, M. Anxiety and depressive disorders in people with epilepsy: A meta-analysis. Epilepsia 2017, 58, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.S.; Sander, J.W.; Taylor, R.J.; Baker, G.A. Predictors of health-related quality of life and costs in adults with epilepsy: A systematic review. Epilepsia 2011, 52, 2168–2180. [Google Scholar] [CrossRef]
- Quek, A.M.L. Autoimmune Epilepsy. Arch. Neurol. 2012, 69, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Bi, M.; Murchison, A.G.; Makuch, M.; Bien, C.G.; Chu, K.; Farooque, P.; Gelfand, J.M.; Geschwind, M.D.; Hirsch, L.J.; et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 2017, 141, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Dubey, D.; Pittock, S.J.; McKeon, A. Antibody Prevalence in Epilepsy and Encephalopathy score: Increased specificity and applicability. Epilepsia 2019, 60, 367–369. [Google Scholar] [CrossRef] [Green Version]
- de Bruijn, M.A.; Bastiaansen, A.E.M.; Mojzisova, H.; Sonderen, A.; Thijs, R.D.; Majoie, M.J.; Rouhl, R.P.W.; Coevorden-Hameete, M.H.; Vries, J.M.; Lopetegi, A.M.; et al. Antibodies Contributing to Focal Epilepsy Signs and Symptoms Score. Ann. Neurol. 2021, 89, 698–710. [Google Scholar] [CrossRef]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangue, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Wu, H.; Osterhaus, G.; Wei, J.; Davis, K.; Sha, D.; Floor, E.; Hsu, C.-C.; Kopke, R.D.; Wu, J.-Y. Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. USA 2003, 100, 4293–4298. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, E.; Dalmau, J. Neuronal autoantigens—Pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 2012, 8, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic Roles of Glutamic Acid Decarboxylase 65 Autoantibodies in Cerebellar Ataxias. J. Immunol. Res. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Falip, M.; Carreño, M.; Miró, J.; Saiz, A.; Villanueva, V.; Quílez, A.; Molins, A.; Barceló, I.; Sierra, A.; Graus, F. Prevalence and immunological spectrum of temporal lobe epilepsy with glutamic acid decarboxylase antibodies. Eur. J. Neurol. 2012, 19, 827–833. [Google Scholar] [CrossRef]
- Carreño, M.; Bien, C.G.; Asadi-Pooya, A.A.; Sperling, M.; Marusic, P.; Elisak, M.; Pimentel, J.; Wehner, T.; Mohanraj, R.; Uranga, J.; et al. Epilepsy surgery in drug resistant temporal lobe epilepsy associated with neuronal antibodies. Epilepsy Res. 2016, 129, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Malter, M.P.; Helmstaedter, C.; Urbach, H.; Vincent, A.; Bien, C.G. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann. Neurol. 2009, 67, 470–478. [Google Scholar] [CrossRef]
- Widman, G.; Golombeck, K.; Hautzel, H.; Gross, C.; Quesada, C.M.; Witt, J.-A.; Rota-Kops, E.; Ermert, J.; Greschus, S.; Surges, R.; et al. Treating a GAD65 Antibody-Associated Limbic Encephalitis with Basiliximab: A Case Study. Front. Neurol. 2015, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Espay, A.J.; Chen, R. Rigidity and spasms from autoimmune encephalomyelopathies: Stiff-person syndrome. Muscle Nerve 2006, 34, 677–690. [Google Scholar] [CrossRef]
- Levy, L.M.; Dalakas, M.C.; Floeter, M.K. The Stiff-Person Syndrome: An Autoimmune Disorder Affecting Neurotransmission of γ-Aminobutyric Acid. Ann. Intern. Med. 1999, 131, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Ali, F.; Rowley, M.; Jayakrishnan, B.; Teuber, S.; Gershwin, M.E.; Mackay, I.R. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: Protean additions to the autoimmune central neuropathies. J. Autoimmun. 2011, 37, 79–87. [Google Scholar] [CrossRef]
- Murinson, B.B.; Guarnaccia, J.B. Stiff-person syndrome with amphiphysin antibodies: Distinctive features of a rare disease. Neurology 2008, 71, 1955–1958. [Google Scholar] [CrossRef]
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.-M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Sharma, C.M.; Kumawat, B.L.; Khandelwal, D.; Gandhi, P. A unique combination of autoimmune limbic encephalitis, type 1 diabetes, and Stiff person syndrome associated with GAD-65 antibody. Ann. Indian Acad. Neurol. 2016, 19, 146–149. [Google Scholar] [CrossRef]
- Seneviratne, S.O.; Buzzard, K.A.; Cruse, B.; Monif, M. Cerebellar Ataxia Followed by Stiff Person Syndrome in a Patient with Anti-GAD Antibodies. Case Rep. Immunol. 2020, 2020, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Balint, B.; Mahant, N.; Meinck, H.-M.; Fung, V. Stiff Limb Syndrome Mimicking Corticobasal Syndrome. Mov. Disord. Clin. Pr. 2014, 1, 354–356. [Google Scholar] [CrossRef]
- Esplin, N.E.; Stelzer, J.W.; Legare, T.B.; Ali, S.K. Difficult to Treat Focal, Stiff Person Syndrome of the Left Upper Extremity. Case Rep. Neurol. Med. 2017, 2017, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Gürol, M.E.; Ertas, M.; Hanagasi, H.A.; Sahin, H.A.; Gürsoy, G.; Emre, M. Stiff leg syndrome: Case report. Mov. Disord. 2001, 16, 1189–1193. [Google Scholar] [CrossRef]
- Saiz, A.; Graus, F.; Valldeoriola, F.; Valls-Solé, J.; Tolosa, E. Stiff-leg syndrome: A focal form of stiff-man syndrome. Ann. Neurol. 1998, 43, 400–403. [Google Scholar] [CrossRef]
- Ghezzi, A.; Montanini, R.; Basso, P.; Zaffaroni, M.; Massimo, E.; Cazzullo, C. Epilepsy in Multiple Sclerosis. Eur. Neurol. 1990, 30, 218–223. [Google Scholar] [CrossRef]
- Engelsen, B.A.; Grønning, M. Epileptic seizures in patients with multiple sclerosis. Is the prognosis of epilepsy underestimated? Seizure 1997, 6, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Miller, F.; Korsvik, H. Baclofen in the treatment of staff-man syndrome. Ann. Neurol. 1981, 9, 511–512. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Fujii, M.; Li, M.; Lutfi, B.; Kyhos, J.; McElroy, B. High-Dose Intravenous Immune Globulin for Stiff-Person Syndrome. New Engl. J. Med. 2001, 345, 1870–1876. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Dai, Y.; Han, B.; Peng, J.; Ma, J.; Tang, Q.; Yang, L. A Case of Anti-GAD 65 Autoimmune Encephalitis Associated with Focal Segmental Stiff-Person Syndrome. Brain Sci. 2023, 13, 369. https://doi.org/10.3390/brainsci13020369
Zhang C, Dai Y, Han B, Peng J, Ma J, Tang Q, Yang L. A Case of Anti-GAD 65 Autoimmune Encephalitis Associated with Focal Segmental Stiff-Person Syndrome. Brain Sciences. 2023; 13(2):369. https://doi.org/10.3390/brainsci13020369
Chicago/Turabian StyleZhang, Chen, Yuwei Dai, Binhong Han, Jian Peng, Jie Ma, Qi Tang, and Li Yang. 2023. "A Case of Anti-GAD 65 Autoimmune Encephalitis Associated with Focal Segmental Stiff-Person Syndrome" Brain Sciences 13, no. 2: 369. https://doi.org/10.3390/brainsci13020369
APA StyleZhang, C., Dai, Y., Han, B., Peng, J., Ma, J., Tang, Q., & Yang, L. (2023). A Case of Anti-GAD 65 Autoimmune Encephalitis Associated with Focal Segmental Stiff-Person Syndrome. Brain Sciences, 13(2), 369. https://doi.org/10.3390/brainsci13020369