Myelin Pathology: Involvement of Molecular Chaperones and the Promise of Chaperonotherapy

 ,
, 
 and
and 
Abstract
:1. Introduction
2. The Structure of Hsp60 and CCT Chaperonins
3. Mutations in the hsp60 Gene
4. Mutation in the CCT5 Subunit Gene
5. Chaperonotherapy
6. Conclusions and Perspectives for the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macario, A.J.L.; Conway de Macario, E. Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Surgucheva, I.; Ninkina, N.; Buchman, V.L.; Grasing, K.; Surguchov, A. Protein aggregation in retinal cells and approaches to cell protection. Cell. Mol. Neurobiol. 2005, 25, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.E.; Metcalf, M.C.; Best, D.; Fleet, G.W.; Garman, S.C. Pharmacological chaperones for human α-N-acetylgalactosaminidase. Proc. Natl. Acad. Sci. USA 2012, 109, 17400–17405. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Fernandez-Guerra, P. Disease-associated mutations in the HSPD1 gene encoding the large subunit of the mitochondrial HSP60/HSP10 chaperonin complex. Front. Mol. Biosci. 2016, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Ouazzani, R.; Chkili, T.; Yahyaoui, M. Autosomal recessive mutilating sensory neuropathy with spastic paraplegia maps to chromosome 5p15.31–14.1. Eur. J Hum. Genet. 2006, 14, 249–252. [Google Scholar] [CrossRef]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Chkili, T.; Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006, 43, 441–443. [Google Scholar] [CrossRef]
- Rangaraju, S.; Hankins, D.; Madorsky, I.; Madorsky, E.; Lee, W.H.; Carter, C.S.; Leeuwenburgh, C.; Notterpek, L. The molecular architecture of myelinated peripheral nerves is supported by calorie restriction with aging. Aging Cell 2009, 8, 178–191. [Google Scholar] [CrossRef]
- de Monasterio-Schrader, P.; Jahn, O.; Tenzer, S.; Wichert, S.P.; Patzig, J.; Werner, H.B. Systematic approaches to central nervous system myelin. Cell. Mol. Life Sci. 2012, 69, 2879–2894. [Google Scholar] [CrossRef]
- Cuéllar, J.; Martín-Benito, J.; Scheres, S.H.W.; Sousa, R.; Moro, F.; López-Viñas, E.; Gómez-Puertas, P.; Muga, A.; Carrascosa, J.L.; Valpuesta, J.M. The structure of CCT–Hsc70NBD suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat. Struct. Mol. Biol. 2008, 15, 858–864. [Google Scholar] [CrossRef]
- Edvardson, S.; Kose, S.; Jalas, C.; Fattal-Valevski, A.; Watanabe, A.; Ogawa, Y.; Mamada, H.; Fedick, A.M.; Ben-Shachar, S.; Treff, N.R.; et al. Leukoencephalopathy and early death associated with an Ashkenazi-Jewish founder mutation in the Hikeshi gene. J. Med. Genet. 2016, 53, 132–137. [Google Scholar] [CrossRef]
- Fontaine, B.; Davoine, C.S.; Durr, A.; Paternotte, C.; Feki, I.; Weissenbach, J.; Hazan, J.; Brice, A. A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q34. Am. J. Hum. Genet. 2000, 66, 702–707. [Google Scholar] [CrossRef]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Heno, B.; et al. Mitochondrial Hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef]
- Hansen, J.; Svenstrup, K.; Ang, D.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Nielsen, J.; Georgopoulos, C.; Bross, P. A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 2007, 254, 897–900. [Google Scholar] [CrossRef]
- Kusk, M.S.; Damgaard, B.; Risom, L.; Hansen, B.; Ostergaard, E. Hypomyelinating leukodystrophy due to HSPD1 mutations: A new patient. Neuropediatrics 2016, 47, 332–335. [Google Scholar] [CrossRef]
- Milner, R.J.; Lai, C.; Nave, K.-A.; Lenoir, D.; Ogata, J.; Sutcliffe, J.G. Nucleotide sequences of two mRNAs for rat brain myelin proteolipid protein. Cell 1985, 42, 931–939. [Google Scholar] [CrossRef]
- Bross, P.; Magnoni, R.; Bie, A.S. Molecular chaperone disorders: Defective Hsp60 in neurodegeneration. Curr. Top. Med. Chem. 2013, 12, 2491–2503. [Google Scholar] [CrossRef]
- Horwich, A.; Fenton, W.; Chapman, E. Two families of chaperonin: Physiology and mechanism. Annu. Rev. Cell Dev. Biol. 2007, 23, 115–145. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006, 6, 65–76. [Google Scholar] [CrossRef]
- Blackstone, C. Hereditary spastic paraplegia. Handb. Clin. Neurol. 2018, 148, 633–652. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Eguchi, T.; Kawahara, K.; Hasegawa, N.; Nakamura, K.; Funakoshi-Tago, M.; Tanoue, A.; Tamura, H.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutation in HSPD1 blunts mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2015, 462, 275–281. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Megumi, F.T.; Hasegawa, N.; Eguchi, T.; Tanoue, A.; Tamura, H.; Yamauchi, J. Data supporting mitochondrial morphological changes by SPG13-associated HSPD1 mutants. Data Brief 2016, 6, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.J.; Dürr, A.; Cournu-Rebeix, I.; Georgopoulos, C.; Ang, D.; Nielsen, M.N.; Davoine, C.S.; Brice, A.; Fontaine, B.; Gregersen, N.; et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002, 70, 1328–1332. [Google Scholar] [CrossRef]
- Hansen, J.J.; Bross, P.; Westergaard, M.; Nielsen, M.N.; Eiberg, H.; Børglum, A.D.; Mogensen, J.; Kristiansen, K.; Bolund, L.; Gregersen, N. Genomic structure of the human mitochondrial chaperonin genes: HSP60 and HSP10 are localised head to head on chromosome 2 separated by a bidirectional promoter. Hum. Genet. 2002, 112, 71–77. [Google Scholar] [CrossRef]
- Bross, P.; Naundrup, S.; Hansen, J.; Nielsen, M.N.; Christensen, J.H.; Kruhøffer, M.; Palmfeldt, J.; Corydon, T.J.; Gregersen, N.; Ang, D.; et al. The Hsp60-(p. V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 2008, 283, 15694–15700. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Kawahara, K.; Torii, T.; Yamauchi, J. Defective myelination in mice harboring hypomyelinating leukodystrophy-associated HSPD1 mutation. Mol. Genet. Metab. Rep. 2017, 11, 6–7. [Google Scholar] [CrossRef]
- Min, W.; Angileri, F.; Luo, H.; Lauria, A.; Shanmugasundaram, M.; Almerico, A.M.; Cappello, F.; Conway de Macario, E.; Lednev, I.K.; Macario, A.J.L.; et al. A human CCT5 gene mutation causing distal neuropathy impairs hexadecamer assembly in an archaeal model. Sci. Rep. 2014, 4, 6688. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, O.A.; Tran, M.T.; Haase-Pettingell, C.; King, J.A. Biochemical characterization of mutants in chaperonin proteins CCT4 and CCT5 associated with hereditary sensory neuropathy. J. Biol. Chem. 2014, 289, 27470–27480. [Google Scholar] [CrossRef]
- Pereira, J.H.; McAndrew, R.P.; Sergeeva, O.A.; Ralston, C.Y.; King, J.A.; Adams, P.D. Structure of the human TRiC/CCT Subunit 5 associated with hereditary sensory neuropathy. Sci. Rep. 2017, 7, 3673. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Stephenson, D.A.; Groves, M.J.; Sweeney, M.G.; Davis, M.B.; An, S.F.; Houlden, H.; Salih, M.A.M.; Timmerman, V.; de Jonghe, P.; et al. Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct4) gene. Hum. Mol. Genet. 2003, 12, 1917–1925. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E.; Cappello, F. The Chaperonopathies. Diseases with Defective Molecular Chaperones; Springer: Dordrecht, The Netherlands; Heidelberg, Germany; New York, NY, USA; London, UK, 2013. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Advances in the understanding and management of neuromuscular diseases. Life Saf. Secur. (LiSS) 2018, 6, 109–118. [Google Scholar]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Rangaraju, S.; Madorsky, I.; Pileggi, J.G.; Kamal, A.; Notterpek, L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol. Dis. 2008, 32, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Shy, M.E.; Balsamo, J.; Lilien, J.; Kamholz, J. A molecular basis for hereditary motor and sensory neuropathy disorders. Curr. Neurol. Neurosci. 2001, 1, 77–88. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef]
- Fortun, J.; Go, J.C.; Li, J.; Amici, S.A.; Dunn, W.A., Jr.; Notterpek, L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol. Dis. 2006, 22, 153–164. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef]
- Campanella, C.; Pace, A.; Bavisotto, C.C.; Marzullo, P.; Gammazza, A.M.; Buscemi, S.; Piccionello, A.P. Heat shock proteins in Alzheimer’s disease: Role and targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef]
- Bajramovic, J.J.; Bsibsi, M.; Geutskens, S.B.; Hassankhan, R.; Verhulst, K.C.; Stege, G.J.J.; de Groot, C.J.A.; van Noort, J.M. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J. Neuroimmunol. 2000, 106, 14–22. [Google Scholar] [CrossRef]
- Carr, A.C.; Khaled, A.S.; Bassiouni, R.; Flores, O.; Nierenberg, D.; Bhatti, H.; Vishnubhotla, P.; Manuel, J.P.; Santra, S.; Khaled, A.R. Targeting chaperonin containing TCP1 (CCT) as a molecular therapeutic for small cell lung cancer. Oncotarget 2017, 8, 110273–110288. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; Fang, F.; Florio, J.B.; Rockenstein, E.; Masliah, E.; Mobley, W.C.; Rissman, R.A.; Wu, C. T-complex protein 1-ring complex enhances retrograde axonal transport by modulating tau phosphorylation. Traffic 2018, 19, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q. Involvement of T-complex protein 1-ring complex/chaperonin containing T-complex protein 1 (TRiC/CCT) in retrograde axonal transport through tau phosphorylation. Neural Regen. Res. 2019, 14, 588–590. [Google Scholar] [CrossRef]



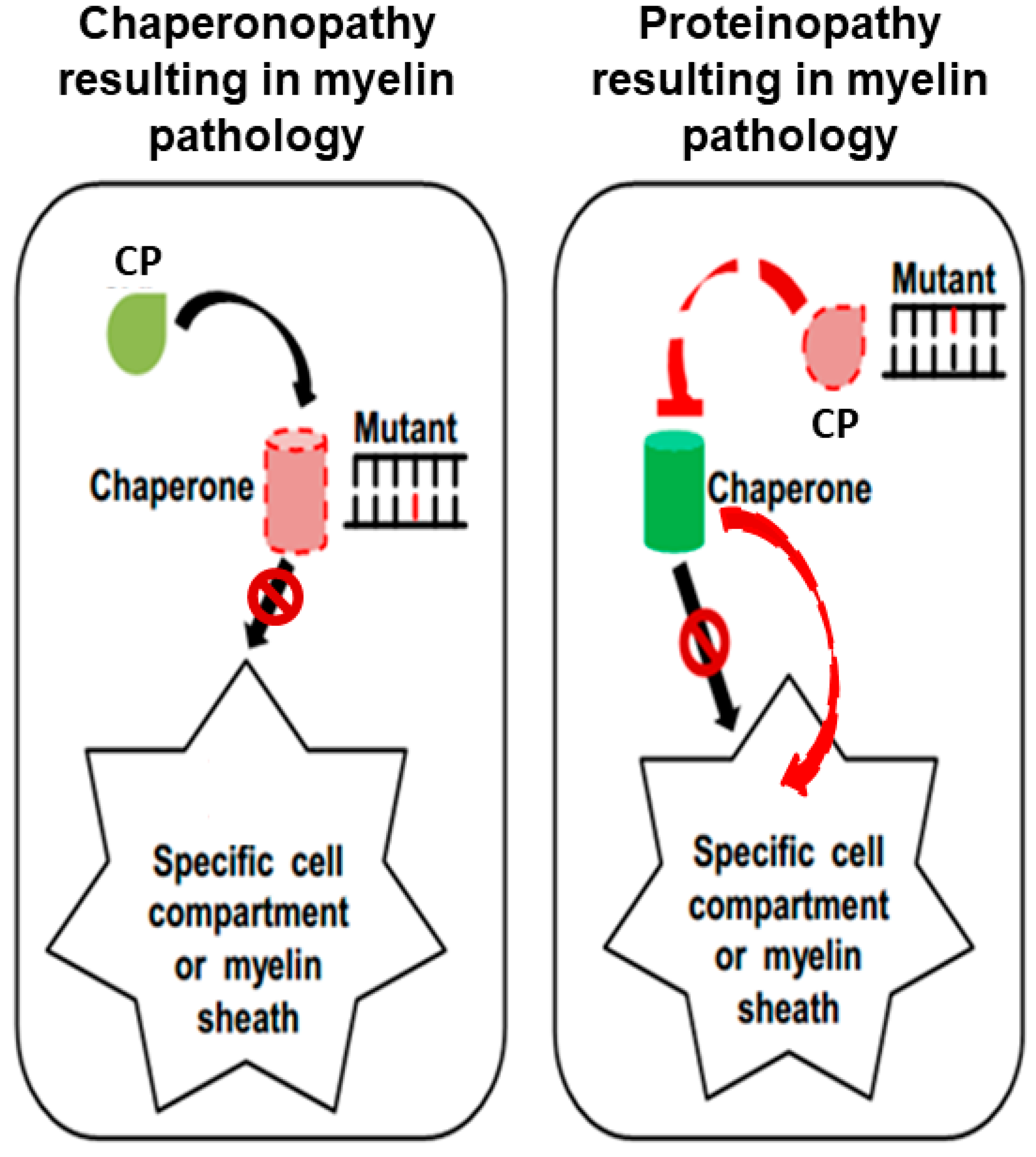{kind=link}
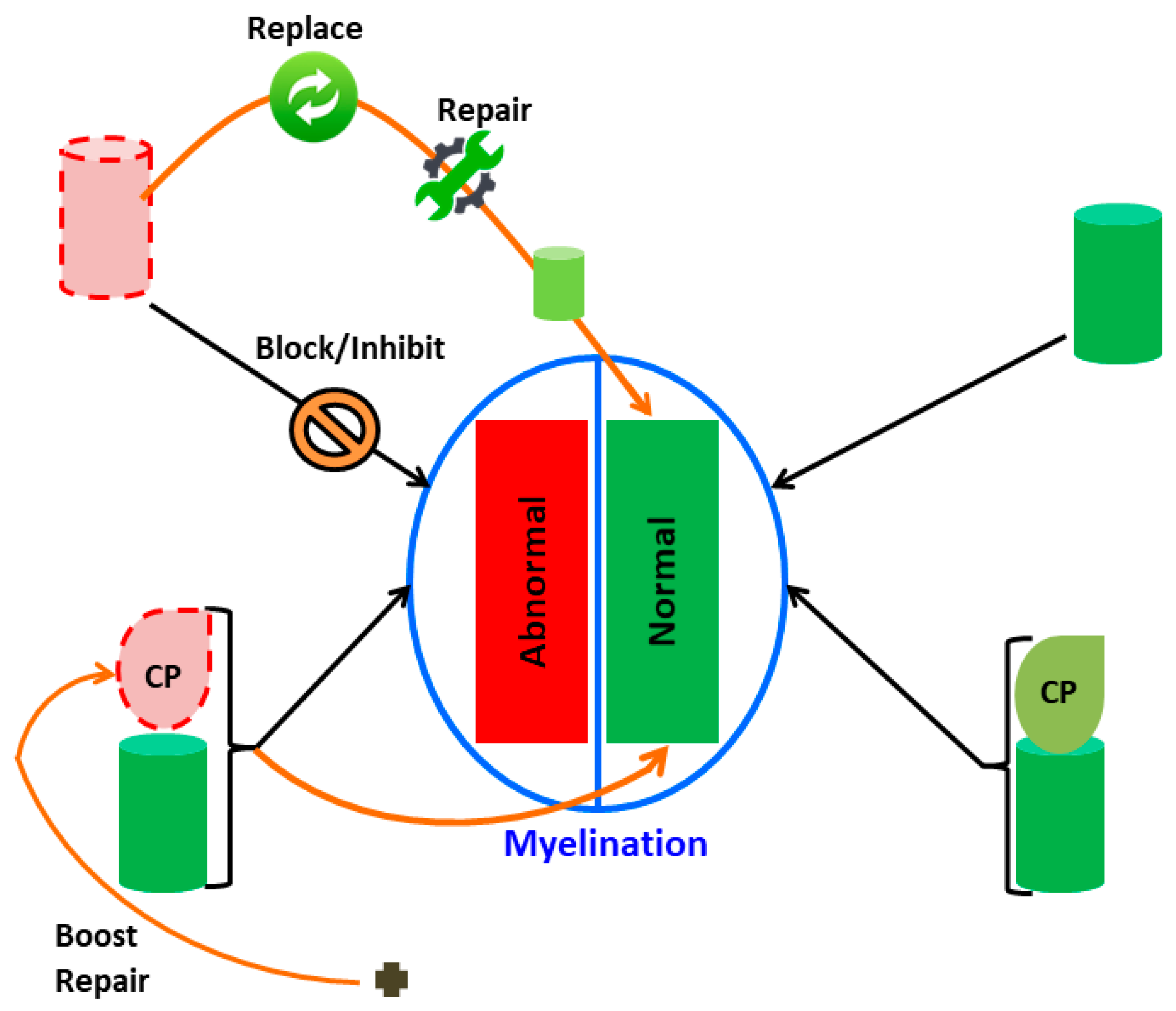{kind=link}
| Disease Myelin Status Chaperonopathy | Mutation | Genetic Condition | Clinical-Pathological Features | Reference |
|---|---|---|---|---|
| Peripheral neuropathy Demyelinating By defect | CCT5-p.His147Arg (consanguineous Moroccan family) | HET | Progressive distal sensory neuropathy of upper and lower limbs leading to mutilating acropathy; abnormality of the lipoprotein profile; severe atrophy of the spinal cord predominantly in the posterior tract (MRI). | [6] |
| SPG13 Possibly demyelinating By defect | Hsp60-p.Val98Ile (French family) | HET 1 | Severe functional handicap; decreased vibration sense; urinary urgency; pes cavus; increased reflexes in the lower and upper limbs; loss of Babinski sign. | [11] |
| MitCHAP-60 disease Hypomyelinating By defect | Hsp60-p.Asp29Gly (consanguineous Israeli Bedouin kindred) | HOM | Rotatory nystagmus, progressive spastic paraplegia; variable rate of neurological deterioration and regression; severe motor impairment; abnormal head control; profound mental retardation; hypomyelinating leukodystrophy (MRI). | [12] |
| Asymptomatic or symptomatic | Hsp60-p.Gln461Glu (Danish HSP patients) | HET | Asymptomatic or symptomatic. Symptomatic cases show: spasticity and weakness in the lower limbs and impaired vibration sense in the toes; normal cerebrum and spinal cord MRI; abnormal motor-evoked and somatosensory evoked potentials; evoked potentials (VEP) abnormal on the left eye. | [13] |
| MitCHAP-60 disease Hypomyelinating By defect | Hsp60-p.Asp29Gly (Syrian boy) | HOM | Slow psychomotor development; absence of heat control; hypotonia; nystagmus; limb spasticity; feeding difficulties; no evidence of normal myelination (MRI) | [14] |
| Chaperonopathy by: | Mechanism, Features | Chaperonotherapy Mode |
|---|---|---|
| Excess | Quantitative, e.g., due to gene dysregulation; upregulation; other. | Negative: Chaperone gene knockdown; inhibition by miRNAs; chaperone-protein blocking (compounds) |
| Qualitative, e.g., gain of function. | Negative: Chaperone gene knockdown; inhibition by miRNAs; chaperone-protein blocking (compounds). | |
| Defect | Quantitative, e.g., gene downregulation; absence or misplacement; sequestration; excessive demand (defect relative to substrate availability); other. | Positive: Chaperone gene/protein replacement; artificial chaperones; chaperone gene induction (e.g., mild harmless stressors); combined. |
| Qualitative, e.g., due to structural defect genetic or acquired (e.g., aberrant post-translational modifications). | Positive: Chaperone gene/protein replacement; artificial chaperones; chaperone function boost (compounds); combined. | |
| Mistake | Normal chaperones (at least as far it can be determined with current methodologies) contribute to disease, e.g., tumors that need chaperones to grow; autoimmune conditions in which a chaperone is the autoantigen and/or induces production of pro-inflammatory cytokines. | Negative: Chaperone gene knockdown; inhibition by miRNAs; chaperone-protein blocking (compounds); combined. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalia, F.; Marino Gammazza, A.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Myelin Pathology: Involvement of Molecular Chaperones and the Promise of Chaperonotherapy. Brain Sci. 2019, 9, 297. https://doi.org/10.3390/brainsci9110297
Scalia F, Marino Gammazza A, Conway de Macario E, Macario AJL, Cappello F. Myelin Pathology: Involvement of Molecular Chaperones and the Promise of Chaperonotherapy. Brain Sciences. 2019; 9(11):297. https://doi.org/10.3390/brainsci9110297
Chicago/Turabian StyleScalia, Federica, Antonella Marino Gammazza, Everly Conway de Macario, Alberto J. L. Macario, and Francesco Cappello. 2019. "Myelin Pathology: Involvement of Molecular Chaperones and the Promise of Chaperonotherapy" Brain Sciences 9, no. 11: 297. https://doi.org/10.3390/brainsci9110297
APA StyleScalia, F., Marino Gammazza, A., Conway de Macario, E., Macario, A. J. L., & Cappello, F. (2019). Myelin Pathology: Involvement of Molecular Chaperones and the Promise of Chaperonotherapy. Brain Sciences, 9(11), 297. https://doi.org/10.3390/brainsci9110297