Effect-Directed Profiling of Powdered Tea Extracts for Catechins, Theaflavins, Flavonols and Caffeine

Abstract
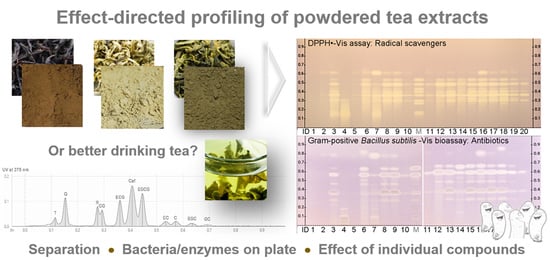
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Stock Solutions, Standard Mixture and Sample Extracts
2.3. Plate Prewashing and Remarks
2.4. RP–HPTLC Method
2.5. Effect–Directed Profiling
- (1)
- DPPH∙ assay: 4 mL 0.04% methanolic DPPH∙ solution was sprayed (green nozzle, level 4). Yellow bands on a purple background were generated instantly. The PC was gallic acid (0.5, 1.3 and 2 µL/band, 0.1 mg/mL in methanol).
- (2)
- Gram-negative A. fischeri bioassay: 150 µL bacterial cryostock were incubated in 20 mL medium according to DIN EN ISO 11348–1 [43] in a 100 mL culture flask at 75 rpm and room temperature for 18–24 h. By shaking the culture flask in a dark room, the green–blue bioluminescence of the bacteria was visually proven to be ready for use, and 4 mL bacterial suspension were sprayed on the plate. The settling down of the vapor was interrupted to transfer the still humid plate to the BioLuminizer cabinet (CAMAG). Fifteen images were recorded over 45 min (exposure time of 60 s, trigger interval 3.0 min). Antibiotics were detected as dark or brightened bands on the instantly bioluminescent plate background. The PC was Caf (0.5, 1.5 and 3 µL/band, 1 mg/mL in methanol).
- (3)
- Gram-positive B. subtilis bioassay, as exception recommended on 10 × 10 cm HPTLC plate silica gel 60 RP–18 WF254s: 3.5 mL bacteria suspension (100 µL bacterial cryostock per 20 mL 2.3% Müller–Hinton broth, optical density of ca. 0.8 at 600 nm [44]) were sprayed (red nozzle), followed by incubation at 37 °C for 2 h. For generation of the colorless (white) antibacterial bands on a purple background, the plate was sprayed with 500 µL 0.2% DPBS-buffered MTT solution and incubated at 37 °C for 45 min, followed by drying (50 °C, 5 min, Plate Heater, CAMAG). The PC was tetracycline (0.5, 1.5 and 3 µL/band, 0.005 mg/mL in ethanol). The application of this bioassay on the LiChrospher® HPTLC plate silica gel 60 RP–18 WF254s required an additional binder hardening by heating the plate at 140 °C for 20 min, a two-fold neutralization of the more acidic layer using a pH 12 buffer (citric acid 6 g/L, di-sodium hydrogen phosphate 10 g/L), a longer (overnight) incubation and higher sample amounts to be applied (as not so sensitive in the detection on this layer).
- (4)
- α-Glucosidase inhibition assay: 2 mL substrate solution (12 mg 4-methylumbelliferyl-α-D-glucopyranoside dissolved in 0.2 mL dimethyl sulfoxide and diluted with 9 mL ethanol and 1 mL 10 mM sodium chloride solution) were sprayed first (green nozzle, level 5), then after drying (2 min), 2.5 mL α-glucosidase solution (10 U/mL in sodium acetate buffer, pH 7.5), followed by incubation (37 °C, 90 min) and drying (3 min). To obtain the most intense 4-methylumbelliferyl-blue fluorescent background, the plate was made alkaline by placing it (15 min) in a dry chamber with the counter trough filled with 10 mL 25% ammonia solution. As contrast, dark inhibition bands absorbing at FLD 366 nm were revealed. The PC was acarbose (1, 3 and 6 µL/band, 3 mg/mL in ethanol).
- (5)
- β-Glucosidase inhibition assay: same as before, but the substrate was 4-methylumbelliferyl-β-D-glucopyranoside, the β-glucosidase solution was 1000 U/mL and the incubation took 90 min. The PC was imidazole (2, 5 and 8 µL/band, 1 mg/mL in ethanol).
- (6)
- Tyrosinase inhibition assay: 2 mL were sprayed each of substrate solution (4.5 mg/mL levodopa in phosphate buffer of 0.14% dipotassium phosphate and 0.16% disodium phosphate, 20 mM, pH 6.8, plus 2.5 mg CHAPS and 7.5 mg PEG 8000), and after drying (1 min), tyrosinase solution (400 U/mL in phosphate buffer), followed by incubation at room temperature for 15–20 min to reveal the colorless (white) inhibition bands on a grey background. The PC was kojic acid (1, 3 and 6 µL/band, 0.1 mg/mL in ethanol).
- (7)
- AChE inhibition assay: The plate was sprayed (green nozzle) with 1.3 mL substrate solution (1 mg/mL indoxyl acetate in ethanol) and then 3 mL AChE solution (6.66 U/mL in Tris–HCl buffer plus 1 mg BSA), followed by incubation at 37 °C for 25 min. Visible indigo-blue inhibition bands were revealed, which were more sensitively detected as absorbing dark bands on the indigo-blue fluorescent plate background at FLD 366 nm. The PC was rivastigmine (2, 4 and 8 µL/band, 0.1 mg/mL in methanol).
- (8)
- β-Glucuronidase inhibition assay: 2.0 mL of β-glucuronidase solution (50 U/mL in potassium phosphate buffer, 0.1 M, pH 7.0) were sprayed (yellow nozzle), followed by incubation at 37 °C for 15 min [45]. For generation of the colorless (white) inhibition bands on an indigo-blue colored background, the plate was sprayed (red nozzle) with 1.5 mL 5-bromo-4-chloro-3-indolyl-β-D-glucuronide solution (2 mg/mL in water) and incubated at 37 °C for 1 h. The PC was D–saccharolactone (0.5, 1.5 and 3 µL/band, 0.1 mg/mL in water).
3. Results and Discussion
3.1. Setup of the Profiling
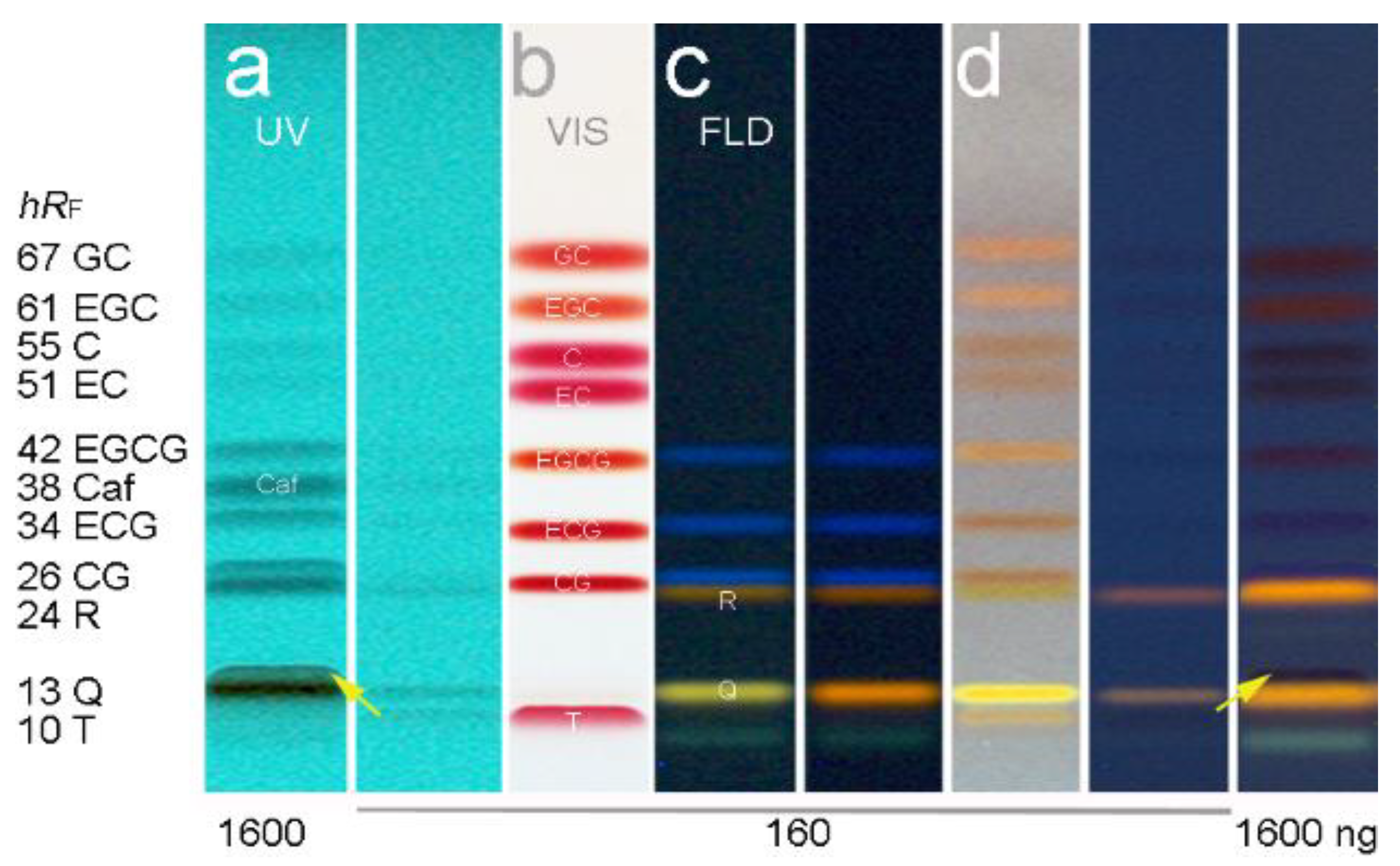
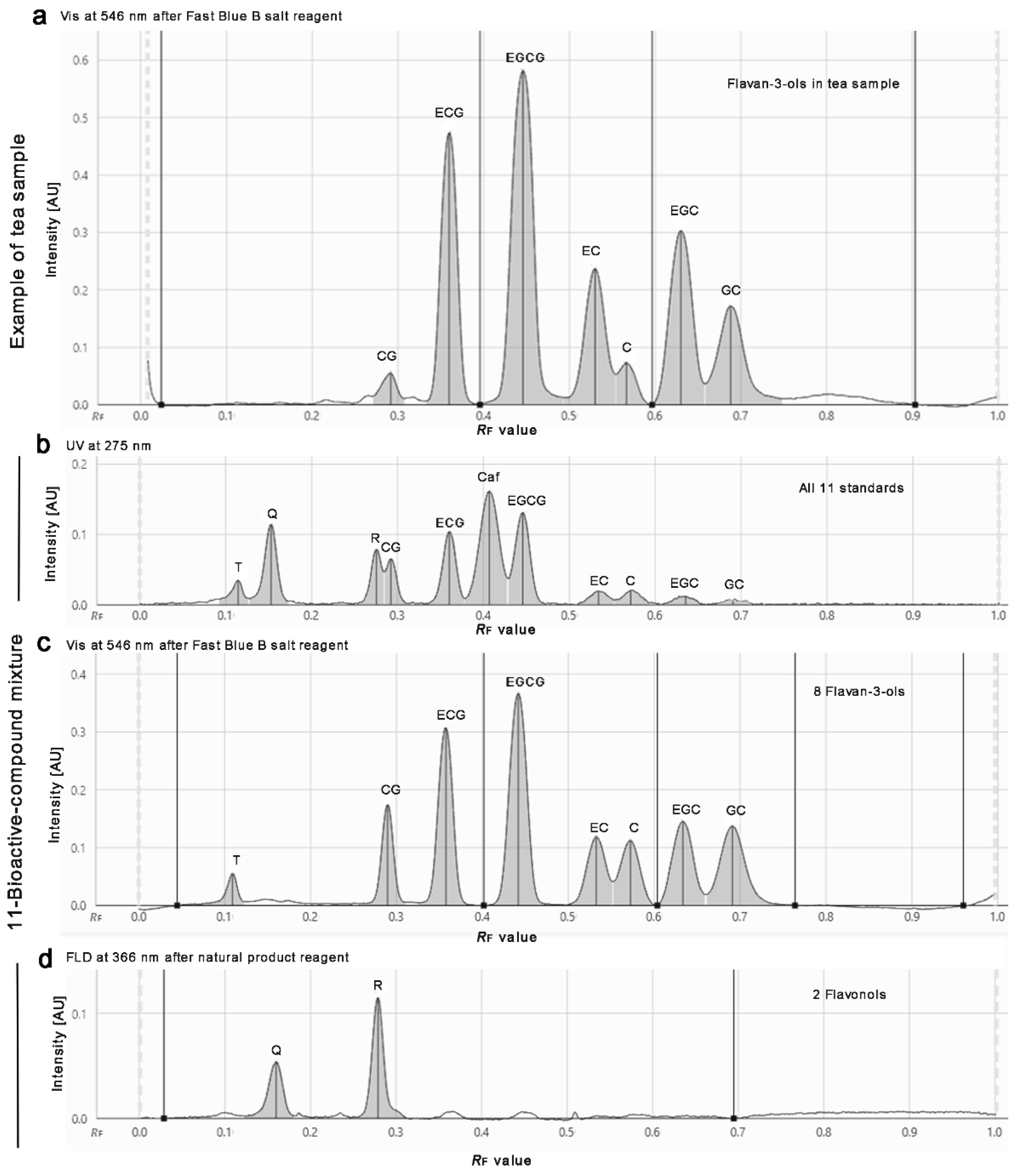
3.2. Method Development
3.3. Development of the Effect-Directed Profiling
3.4. Results of the Effect-Directed Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dietary Supplements Market Size, Share & Trends Analysis Report by Ingredient (Vitamins, Minerals), by Form, by Application, by End User, by Distribution Channel, by Region, and segment forecasts, 2020–2027. Available online: www.grandviewresearch.com/industry-analysis/dietary-supplements-market (accessed on 14 January 2021).
- Stodt, U.; Engelhardt, U.H. Progress in the analysis of selected tea constituents over the past 20 years. Food Res. Int. 2013, 53, 636–648. [Google Scholar] [CrossRef]
- Silva Pinto, M. Tea: A new perspective on health benefits. Food Res. Int. 2013, 53, 558–567. [Google Scholar] [CrossRef]
- EFSA Panel on Food Additives and Nutrient Sources added to Food, (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; et al. Scientific opinion on the safety of green tea catechins. EFSA J. 2018, 16, e05239. [Google Scholar] [PubMed] [Green Version]
- Karwowska, K.; Skotnicka, M.; Smiechowska, M. Tea production and its forecasts, and the possibility of tea cultivation in the context of environmental requirements in China. Sci. J. Wars. Univ. Life Sci. SGGW 2019, 19, 180–191. [Google Scholar]
- Biesterbos, J.W.H.; Sijm, D.T.H.M.; Van Dam, R.; Mol, H.G.J. A health risk for consumers: The presence of adulterated food supplements in the Netherlands. Food Addit. Contam. Part A 2019, 36, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Rocha, T.; Amaral, J.S.; Oliveira, M.B.P.P. Adulteration of Dietary Supplements by the Illegal Addition of Synthetic Drugs: A Review. CRFSFS 2016, 15, 43–62. [Google Scholar] [CrossRef]
- Gilroy, C.M.; Steiner, J.F.; Byers, T.; Shapiro, H.; Georgian, W. Echinacea and truth in labeling. Arch. Intern. Med. 2003, 163, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Sherma, J.; Rabel, F. Advances in the thin layer chromatographic analysis of counterfeit pharmaceutical products: 2008–2019. J. Liq. Chromatogr. Relat. Technol. 2019, 42, 367–379. [Google Scholar] [CrossRef]
- Venditti, E.; Bacchetti, T.; Tiano, L.; Carloni, P.; Greci, L.; Damiani, E. Hot vs. cold water steeping of different teas: Do they affect antioxidant activity? Food Chem. 2010, 119, 1597–1604. [Google Scholar] [CrossRef]
- Bundeszentrum für Ernährung, BZfE (2019): Tee: Einkauf und Kennzeichnung. Available online: www.bzfe.de/inhalt/tee-einkauf-und-kennzeichnung-28489.html (accessed on 14 January 2021).
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [Green Version]
- Dulloo, A.G.; Seydoux, J.; Girardier, L.; Chantre, P.; Vandermander, J. Green tea and thermogenesis: Interactions between catechin-polyphenols, caffeine and sympathetic activity. Int. J. Obes. 2000, 24, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufresne, C.J.; Farnworth, E.R. A review of latest research findings on the health promotion properties of tea. J. Nutr. Biochem. 2001, 12, 404–421. [Google Scholar] [CrossRef]
- Betts, J.W.; Kelly, S.M.; Haswell, S.J. Antibacterial effects of theaflavin and synergy with epicatechin against clinical isolates of Acinetobacter baumannii and Stenotrophomonas maltophilia. Int. J. Antimicrob. Agents 2011, 38, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, M.; Takemoto, H. Synthesis of Theaflavins and Their Functions. Molecules. 2018, 23, 918. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, U.H. 3.23 Chemistry of Tea. In Comprehensive Natural Products II, 1st ed.; Liu, H.W., Mander, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 999–1032. [Google Scholar]
- Matsuo, Y.; Hayashi, T.; Saito, Y.; Kouno, I.; Tanaka, T. Structures of enzymatic oxidation products of epigallocatechin. Tetrahedron 2013, 69, 8952–8958. [Google Scholar] [CrossRef] [Green Version]
- Yassin, G.H.; Koek, J.H.; Kuhnert, N. Model system-based mechanistic studies of black tea thearubigin formation. Food Chem. 2015, 180, 272–279. [Google Scholar] [CrossRef]
- Bansala, S.; Choudhary, S.; Sharma, M.; Kumar, S.S.; Lohan, S.; Bhardwaj, V.; Syan, N.; Jyoti, S. Tea: A native source of antimicrobial agents. Food Res. Int. 2013, 53, 568–584. [Google Scholar] [CrossRef]
- Kusumawardani, A.; Sukmasari, S.; Abdul Mutalib, N.A.; Abdul Rahman, S.F.; Ichwan, S.J.A. Comparative study of antimicrobial potential of White tea and Black tea leaf extracts from East Java–Indonesia on two species of oral streptococci. Mater. Today 2019, 16, 2226–2230. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chakraborti, S. Bioassay-Guides Isolation and Identification of Antibacterial and Antifungal Components from Methanolic Extract of Green Tea Leaves (Camellia sinensis). Res. J. Phytochem. 2010, 4, 78–86. [Google Scholar]
- Yam, T.S.; Shah, S.; Hamilton-Miller, J. Microbiological activity of whole and fractionated crude extracts of tea (Camellia sinensis), and of tea components. FEMS Microbiol. Lett. 1997, 152, 169–174. [Google Scholar] [CrossRef]
- Henning, S.M.; Fajardo-Lira, C.; Lee, H.W.; Youssefian, A.A.; Go, V.L.W.; Heber, D. Catechin content of 18 teas and a green tea extract supplement correlates with the antioxidant capacity. Nutr. Cancer 2003, 45, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.S.; Kim, S.H.; Kim, Y.B.; Kim, Y.B. Quantitative analysis of major constituents in green tea with different plucking periods and their antioxidant activity. Molecules 2014, 19, 9173–9186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, N.; Ishigaki, F.; Ishigaki, A.; Iwashina, H.; Hara, Y. Reduction of Blood Glucose Levels by Tea Catechin. Biosci. Biotechnol. Biochem. 1993, 57, 525–527. [Google Scholar] [CrossRef]
- Mandel, S.; Youdim, M.B.H. Catechin polyphenols: Neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic. Biol. Med. 2004, 37, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Li, H.; Sinha, S.; Bitan, G. Chapter 6—Modulators of Amyloid β-Protein (Aβ) Self-Assembly. In Developing Therapeutics for Alzheimer’s Disease Progress and Challenges; Michael, S., Wolfe, M.S., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2016; pp. 97–191. [Google Scholar]
- Wohlmuth, H.; Savage, K.; Dowell, A.; Mouatt, P. Adulteration of Ginkgo biloba products and a simple method to improve its detection. Phytomedicine 2014, 21, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Li, P.; Zhang, Q.; Zhang, W.; Ding, X. Determination for major chemical contaminants in tea (Camellia sinensis) matrices: A review. Food Res. Int. 2013, 53, 649–658. [Google Scholar] [CrossRef]
- Dayarathna, D.; Thirimanna, C.; Mubarak, A.; Mackay, L.; Rogerson, J.; Tang, H.; Feng, J.; Sin, D.W.M.; Wong, Y.C. A multinational joint project on the evaluation of residual pesticide analysis in tea in the Asia Pacific region. Food Res. Int. 2013, 53, 931–937. [Google Scholar] [CrossRef]
- El-Shahawi, M.S.; Hamza, A.; Bahaffi, S.O.; Al-Sibaai, A.A.; Abduljabbar, T.N. Analysis of some selected catechins and caffeine in green tea by high performance liquid chromatography. Food Chem. 2012, 134, 2268–2275. [Google Scholar] [CrossRef]
- Chen, Q.; Guo, Z.; Zhao, J. Identification of green tea’s (Camellia sinensis (L.)) quality level according to measurement of main catechins and caffeine contents by HPLC and support vector classification pattern recognition. J. Pharm. Biomed. Anal. 2008, 48, 1321–1325. [Google Scholar] [CrossRef]
- Naldi, M.; Fiori, J.; Gotti, R.; Périat, A.; Veuthey, J.L.; Guillarme, D.; Andrisano, V. UHPLC determination of catechins for the quality control of green tea. J. Pharm. Biomed. Anal. 2014, 88, 307–314. [Google Scholar] [CrossRef]
- Glavnik, V.; Simonovska, B.; Vovk, I. Densitometric determination of (+)-catechin and (−)-epicatechin by 4-dimethylaminocinnamaldehyde reagent. J. Chromatogr. A 2009, 1216, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Z.; Huang, J.A.; Fu, D.; Liu, F.; Gong, Y.; Wu, X. TLC separation of catechins and theaflavins on polyamide plates. J. Planar Chromatogr. 2009, 22, 97–100. [Google Scholar] [CrossRef]
- Abd-ElSalam, H.A.H.; Al-Ghobashy, M.A.; Zaazaa, H.E.; Ibrahim, M.A. Stability of catechins in green tea nutraceutical products: Application of solid phase extraction-thin layer chromatography densitometry. Food Chem. 2014, 156, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Baba, W.N.; Gani, A.; Wani, T.A.; Gani, A.; Masoodi, F.A. Effect of extraction time on antioxidants and bioactive volatile components of green tea (Camellia sinensis), using GC/MS. Cogent Food Agric. 2015, 1, 1106387. [Google Scholar] [CrossRef]
- Yang, Z.; Baldermann, S.; Watanabe, N. Recent studies of the volatile compounds in tea. Food Res. Int. 2013, 53, 585–599. [Google Scholar] [CrossRef]
- Vovk, I.; Simonovska, B.; Vuorela, H.J. Separation of eight selected flavan-3-ols on cellulose thin-layer chromatographic plates. J. Chromatogr. A 2005, 1077, 188–194. [Google Scholar] [CrossRef]
- Reich, E.; Schibli, A.; Widmer, V.; Jorns, R.; Wolfram, E.; DeBatt, A. HPTLC Methods for Identification of Green Tea and Green Tea Extract. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 2141–2151. [Google Scholar] [CrossRef]
- DIN EN ISO. 11348–1: Water Quality—Determination of the Inhibitory Effect of Water Samples on the Light Emission of Vibrio Fischeri (Luminescent Bacteria Test)—Part 1: Method Using Freshly Prepared Bacteria; Beuth Verlag: Berlin, Germany, 2009. [Google Scholar]
- Jamshidi-Aidji, M.; Morlock, G.E. From bioprofiling and characterization to bioquantification of natural antibiotics by direct bioautography linked to high-resolution mass spectrometry: Exemplarily shown for Salvia miltiorrhiza root. Anal. Chem. 2016, 88, 10979–10986. [Google Scholar] [CrossRef]
- Mahran, E.; Keusgen, M.; Morlock, G.E. New planar assay for a streamlined detection and quantification of β-glucuronidase inhibitors and application to botanical extracts. Anal. Chim. Acta X 2020, 4, 100039. [Google Scholar]
- Morlock, G. Background mass signals in TLC/HPTLC–ESI–MS and practical advices for use of the TLC–MS Interface. J. Liq. Chromatogr. Relat. Technol. 2014, 37, 2892–2914. [Google Scholar] [CrossRef]
- Morlock, G. Chapter 49: High-Performance Thin-Layer Chromatography–Mass Spectrometry for Analysis of Small Molecules. In Mass Spectrometry Handbook; Lee, M., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 1181–1206. [Google Scholar]
- Azadniya, E.; Morlock, G.E. Automated piezoelectric spraying of biological and enzymatic assays for effect-directed analysis of planar chromatograms. J. Chromatogr. A 2019, 1602, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Pobłocka-Olech, L.; Krauze-Baranowska, M.; Wiwart, M. HPTLC determination of catechins in different clones of the genus Salix. J. Planar Chromatogr. 2007, 20, 61–64. [Google Scholar] [CrossRef]
- Glavnik, V.; Vovk, I. High performance thin-layer chromatography–mass spectrometry methods on diol stationary phase for the analyses of flavan-3-ols and proanthocyanidins in invasive Japanese knotweed. J. Chromatogr. A 2019, 1598, 196–208. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morlock, G.E.; Heil, J.; Inarejos-Garcia, A.M.; Maeder, J. Effect-Directed Profiling of Powdered Tea Extracts for Catechins, Theaflavins, Flavonols and Caffeine. Antioxidants 2021, 10, 117. https://doi.org/10.3390/antiox10010117
Morlock GE, Heil J, Inarejos-Garcia AM, Maeder J. Effect-Directed Profiling of Powdered Tea Extracts for Catechins, Theaflavins, Flavonols and Caffeine. Antioxidants. 2021; 10(1):117. https://doi.org/10.3390/antiox10010117
Chicago/Turabian StyleMorlock, Gertrud E., Julia Heil, Antonio M. Inarejos-Garcia, and Jens Maeder. 2021. "Effect-Directed Profiling of Powdered Tea Extracts for Catechins, Theaflavins, Flavonols and Caffeine" Antioxidants 10, no. 1: 117. https://doi.org/10.3390/antiox10010117
APA StyleMorlock, G. E., Heil, J., Inarejos-Garcia, A. M., & Maeder, J. (2021). Effect-Directed Profiling of Powdered Tea Extracts for Catechins, Theaflavins, Flavonols and Caffeine. Antioxidants, 10(1), 117. https://doi.org/10.3390/antiox10010117