Anti-Tumor Activity of Hypericum perforatum L. and Hyperforin through Modulation of Inflammatory Signaling, ROS Generation and Proton Dynamics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Signaling Pathways in Cancer and Molecular Targets Susceptible to Therapeutic Intervention by Phytochemicals
2.1. Oncogenesis
2.2. Tumor Growth, Angiogenesis, Invasiveness, and Metastasis
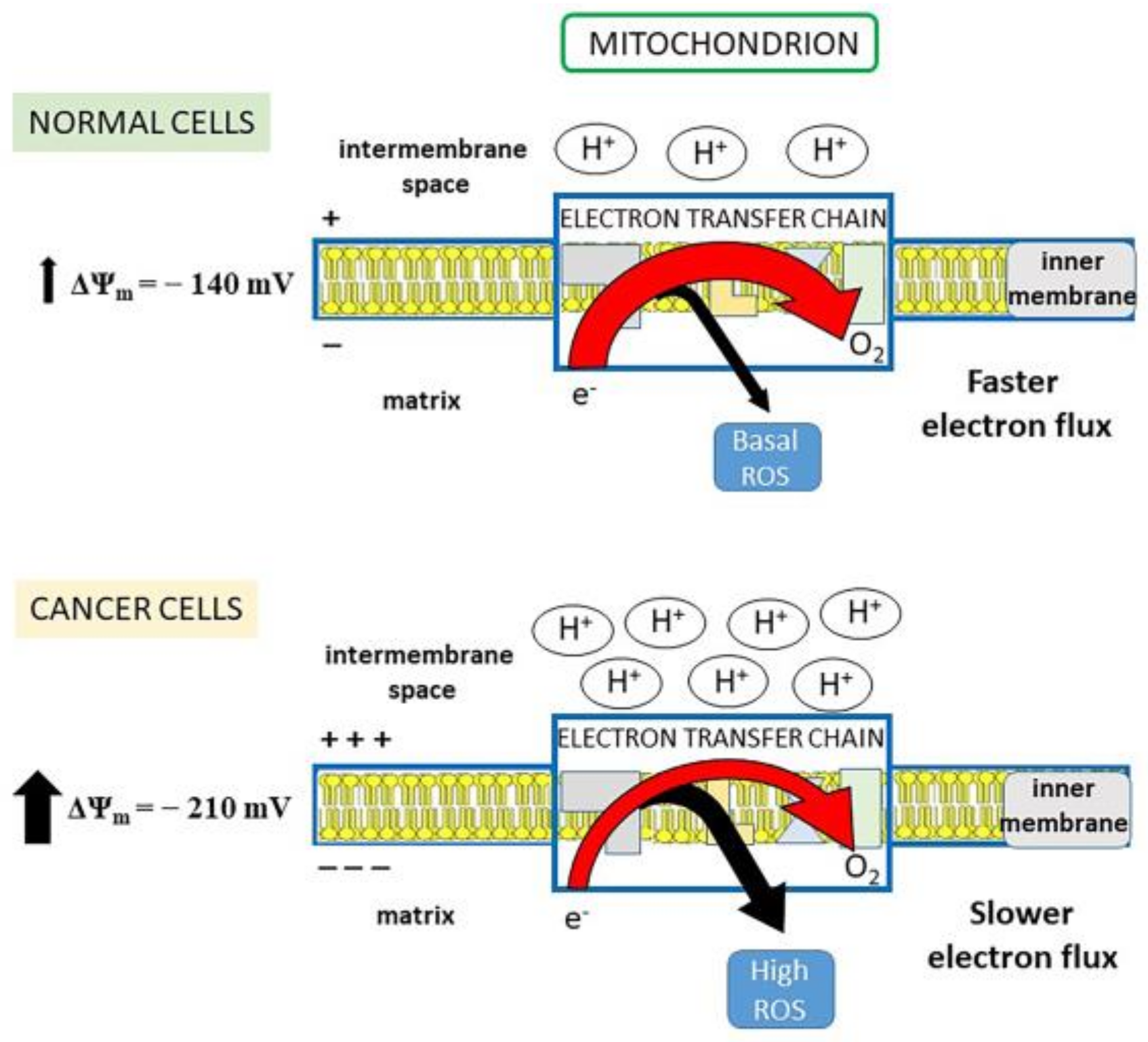
3. Reactive Oxygen Species as a Double-Edged Sword in the Fight against Cancer
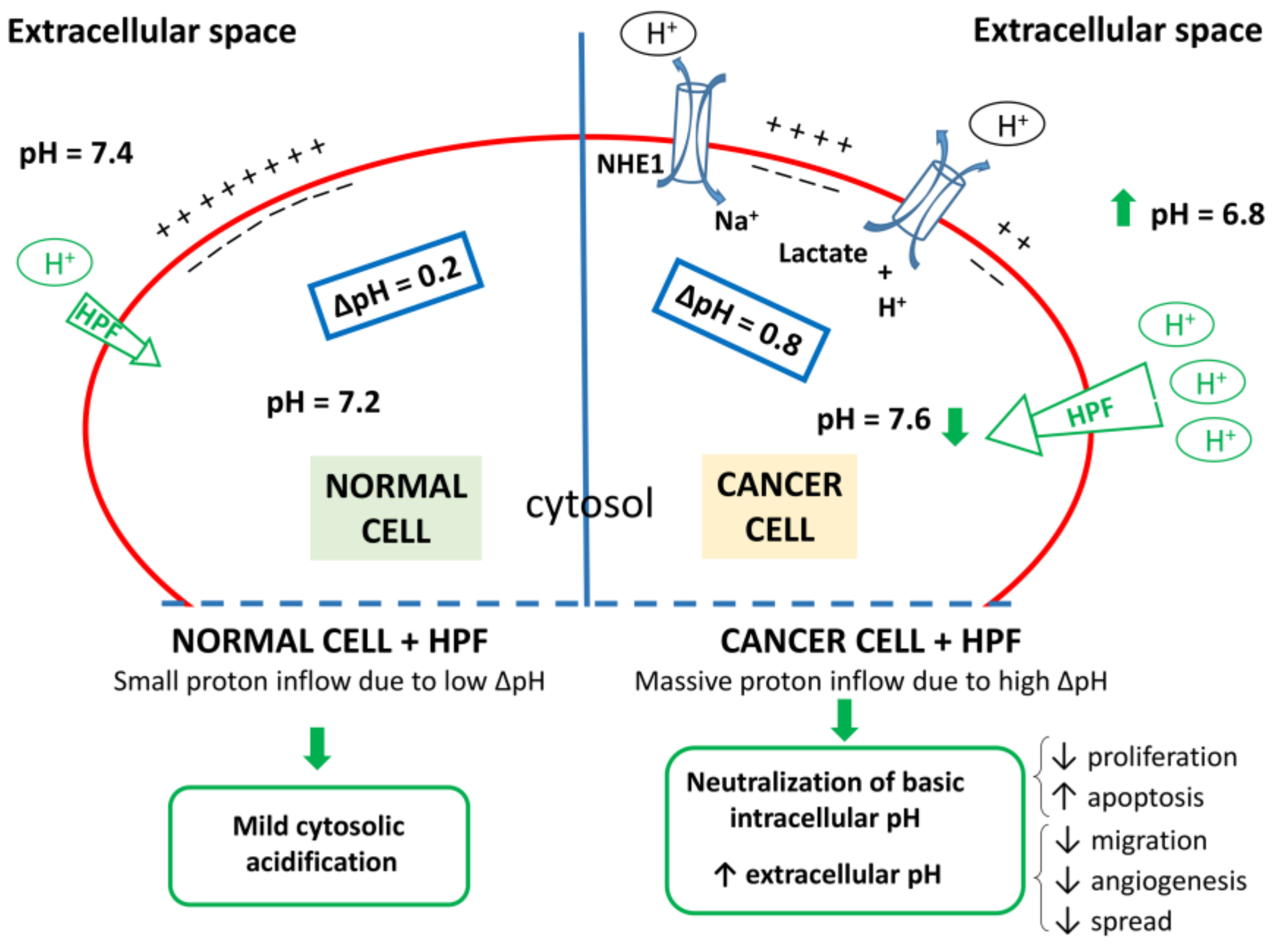
4. Dysregulation of Proton and Ion Content in Cancer Cells as a Possible Target for Phytochemical Therapy
5. Hypericum perforatum or St. John’s Wort (SJW) and Hyperforin (HPF)
5.1. Protective Effects of SJW and HPF against Noxious Stimuli
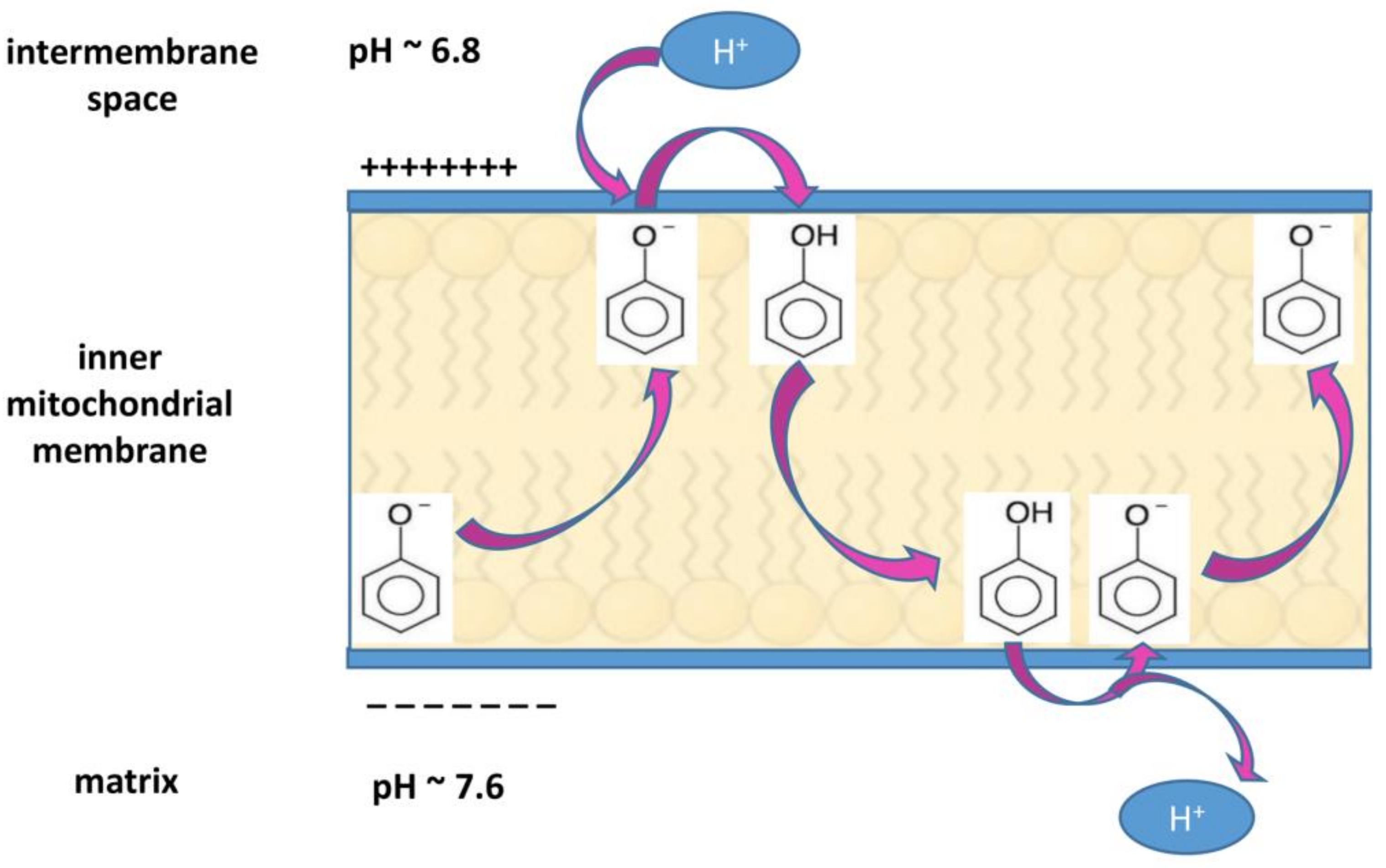
5.2. Molecular Mechanisms of HPF Related to Its Protonophore Activity
6. Antitumor Activity of SJW Extract and Its Component HPF
6.1. Protective Effects of SJW and HPF against Carcinogenesis
6.2. Effects of SJW and HPF on Cell Proliferation and Apoptosis
6.3. SJW and HPF Affect Neo-Angiogenesis, Tumor Cell Invasion and Metastasis.
7. Conclusions
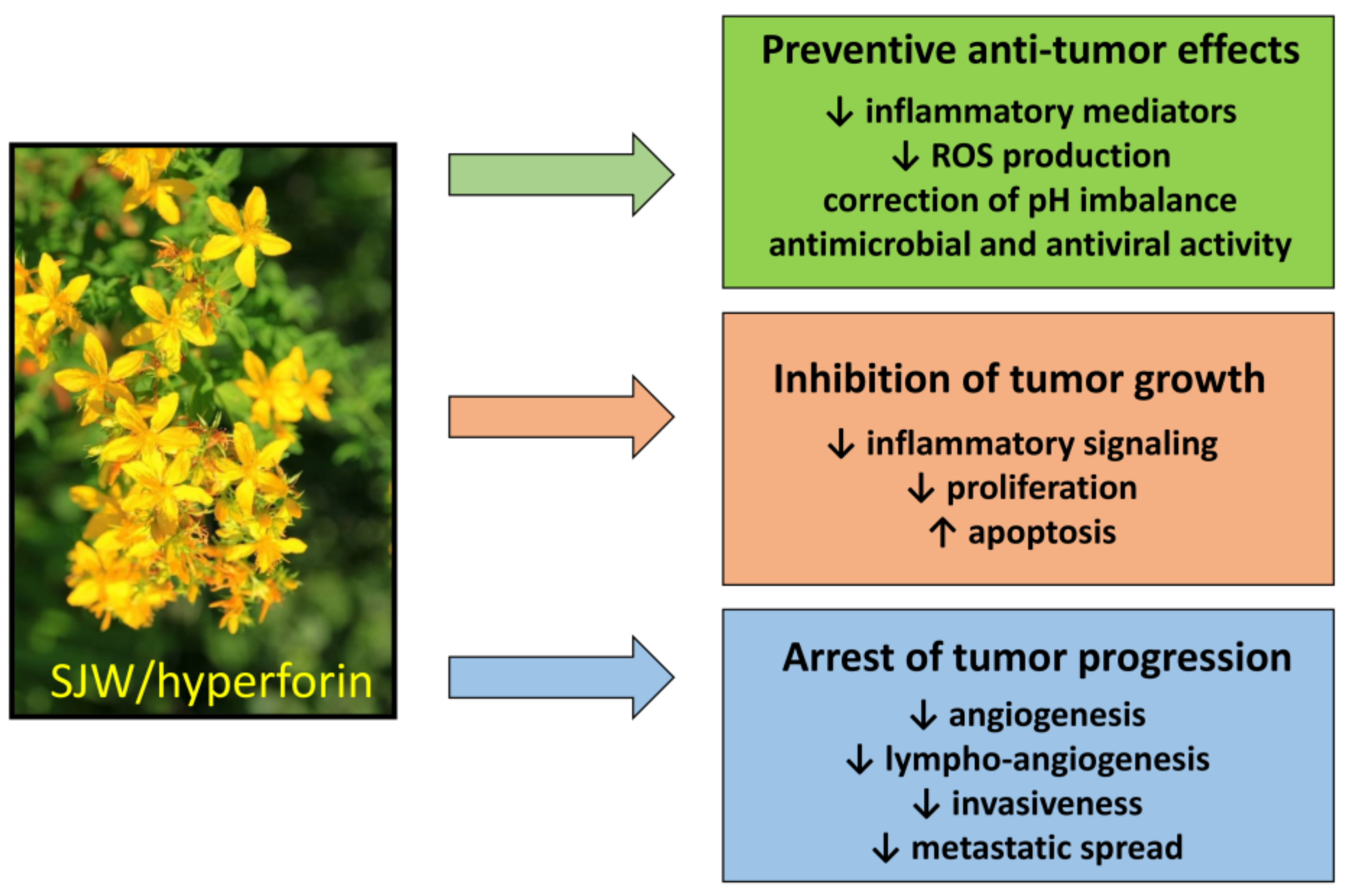
- Tumor prevention. Regular use of SJW reduces cancer risk, by preventing the genotoxic effect of carcinogens [144]. This protective action is essentially based on the ability of HPF to slowdown inflammatory mediators and regulate ROS production and/or pH imbalance, resulting in counteraction of malignant phenotype. It should be emphasized that preventive dietary supplementation of SJW has been found to reduce the risk of colorectal cancer also in humans [156].
- Effects on tumor growth and spread. The anti-inflammatory and anti-angiogenic effects of SJW/HPF, presumably due to inhibition of cytokine and chemokine production and hindrance of their downstream signaling, can protect from tumor expansion. Additionally, the protonophore property of HPF can avoid the acidification of the tumor extracellular milieu, impairing neo-angiogenesis and metalloproteinases activity and thereby tumor invasiveness and metastatic spread [116]. The HPF ability to drop off mitochondrial membrane hyperpolarization and consequently mtROS generation inhibits cell proliferation and favors apoptosis induction. Indeed, the pro-apoptotic effect of SJW/HPF is well documented in many malignant cell lines or in animal tumor models and appears determined by the imbalance between pro- and anti-apoptotic protein expression, even though the concentrations of HPF capable of inducing apoptosis by such mechanism are quite high (at least 5–10 µM) and difficult to be achieved in clinical treatment. Nevertheless, it is well known that proliferating cancer cells display higher Δψm than normal cells, and that positive correlation exists between the malignancy grade of cell clones and their mitochondrial potential [195]. Consequently, malignant cells are more sensitive than normal cells to even small changes of the mitochondrial electro-chemical gradient. Thus, low doses of SJW/HPF, likely insufficient to induce cancer cell death, can nevertheless block ROS-elicited tumor growth and spread by hindering activity of pro-survival protein kinases and angiogenesis.
- Bioavailability and broad spectrum of action. Furthermore, SJW/HPF, for their large bioavailability, persistence of protective benefits and substantial absence of adverse effects, are natural products of biological relevance for tumor prevention and treatment. As cancer therapy requires a multifactorial strategy, SJW/HPF treatment can actually meet this requirement, due to their pleiotropic effects against many different molecular targets along signaling pathways crucial for tumor growth and progression. Additionally, SJW/HPF, acting against different types of tumor cells, can be a broad-spectrum anti-tumor compound and should be tested in association with current chemotherapy drugs to achieve additive effects.
- Limitations. The therapeutic use of HPF-containing SJW extract as anti-depressant has confirmed its very good tolerability and the paucity of adverse effects. However, a major concern of SJW/HPF treatment regards the possible occurrence of drug-drug interaction, due to HPF high affinity binding to PXR, resulting in increased expression levels of cytochrome P450 isoenzymes [141]. Actually, by activating CYP3A4, HPF can enhance drug metabolism and excretion, thereby reducing the effectiveness of a number of chemotherapeutic agents. Thus, the association of SJW/HPF to a chemotherapeutic drug should be thoughtfully decided and monitored to ensure that the drug efficacy is maintained. Very interestingly, several phloroglucinol derivatives devoid of PXR binding activity, have recently been characterized [196]. These HPF derivatives have already been proven to maintain antidepressant activity, although antitumor properties have not yet been tested and should be investigated in a near future.
Author Contributions
Funding
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharm. 2019, 10, 1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.-J.; Qi, M.; Li, N.; Lei, Y.-H.; Zhang, D.-M.; Chen, J.-X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L. Ins and outs of dietary phytochemicals in cancer chemoprevention. Biochem. Pharm. 2007, 74, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Khor, T.O.; Shu, L.; Su, Z.-Y.; Fuentes, F.; Kong, A.-N.T. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol. Ther. 2013, 137, 153–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surh, Y.-J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, Y.; Zhang, C.; Wu, R.; Yang, A.Y.; Gaspar, J.; Kong, A.-N.T. Dietary Phytochemicals and Cancer Chemoprevention: A Perspective on Oxidative Stress, Inflammation, and Epigenetics. Chem. Res. Toxicol. 2016, 29, 2071–2095. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Borrello, M.G.; Alberti, L.; Fischer, A.; Degl’innocenti, D.; Ferrario, C.; Gariboldi, M.; Marchesi, F.; Allavena, P.; Greco, A.; Collini, P.; et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc. Natl. Acad. Sci. USA 2005, 102, 14825–14830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.; Charles, K.A.; Balkwill, F.R. Tumour necrosis factor-alpha as a tumour promoter. Eur. J. Cancer 2006, 42, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P. Can NF-κB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View. Int. J. Mol. Sci. 2020, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, S.H.M.; Makpol, S.; Abdul Hamid, N.A.; Das, S.; Ngah, W.Z.W.; Yusof, Y.A.M. Ginger extract (Zingiber officinale) has anti-cancer and anti-inflammatory effects on ethionine-induced hepatoma rats. Clinics 2008, 63, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, M.T.; Perduca, M.; Romanelli, M.G.; Mottes, M.; Dalle Carbonare, L. A potential role for astaxanthin in the treatment of bone diseases (Review). Mol. Med. Rep. 2020, 22, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Rho, O.; Junco, J.; Carbajal, S.; Siegel, D.; Slaga, T.J.; DiGiovanni, J. Effect of Combined Treatment with Ursolic Acid and Resveratrol on Skin Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate. Cancer Prev. Res. 2015, 8, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristea, S.; Sage, J. Is the Canonical RAF/MEK/ERK Signaling Pathway a Therapeutic Target in SCLC? J. Thorac. Oncol. 2016, 11, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Ekrami, E.M.; Aghdas, S.A.M.; Mihanfar, A.; Hallaj, S.; Yousefi, B.; Safa, A.; Majidinia, M. Targeting PI3K/Akt/mTOR signaling pathway by polyphenols: Implication for cancer therapy. Life Sci. 2020, 255, 117481. [Google Scholar] [CrossRef] [PubMed]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Rad, E.; Murray, J.T.; Tee, A.R. Oncogenic Signalling through Mechanistic Target of Rapamycin (mTOR): A Driver of Metabolic Transformation and Cancer Progression. Cancers 2018, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Hu, Y.-C.; Dong, S.; Fan, M.; Tamae, D.; Ozeki, M.; Gao, Q.; Gius, D.; Li, J.J. Co-activation of ERK, NF-kappaB, and GADD45beta in response to ionizing radiation. J. Biol. Chem. 2005, 280, 12593–12601. [Google Scholar] [CrossRef] [Green Version]
- Kucharczak, J.; Simmons, M.J.; Fan, Y.; Gélinas, C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene 2003, 22, 8961–8982. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Mahalanobish, S.; Saha, S.; Ghosh, S.; Sil, P.C. Natural products: An upcoming therapeutic approach to cancer. Food Chem. Toxicol. 2019, 128, 240–255. [Google Scholar] [CrossRef]
- Stevens, J.F.; Revel, J.S.; Maier, C.S. Mitochondria-Centric Review of Polyphenol Bioactivity in Cancer Models. Antioxid. Redox Signal. 2018, 29, 1589–1611. [Google Scholar] [CrossRef]
- Kumar, M.; Kaur, V.; Kumar, S.; Kaur, S. Phytoconstituents as apoptosis inducing agents: Strategy to combat cancer. Cytotechnology 2016, 68, 531–563. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubair, H.; Khan, M.A.; Anand, S.; Srivastava, S.K.; Singh, S.; Singh, A.P. Modulation of the tumor microenvironment by natural agents: Implications for cancer prevention and therapy. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.-Q.; Du, W.-L.; Cai, M.-H.; Yao, J.-Y.; Zhao, Y.-Y.; Mou, X.-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. Mast cells, angiogenesis, and tumour growth. Biochim. Biophys. Acta (Bba) Mol. Basis Dis. 2012, 1822, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Kapinova, A.; Kubatka, P.; Liskova, A.; Baranenko, D.; Kruzliak, P.; Matta, M.; Büsselberg, D.; Malicherova, B.; Zulli, A.; Kwon, T.K.; et al. Controlling metastatic cancer: The role of phytochemicals in cell signaling. J. Cancer Res. Clin. Oncol. 2019, 145, 1087–1109. [Google Scholar] [CrossRef]
- Choi, Y.K.; Cho, S.-G.; Woo, S.-M.; Yun, Y.J.; Park, S.; Shin, Y.C.; Ko, S.-G. Herbal extract SH003 suppresses tumor growth and metastasis of MDA-MB-231 breast cancer cells by inhibiting STAT3-IL-6 signaling. Mediat. Inflamm. 2014, 2014, 492173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Qiu, S.; Liu, P.; Ge, Y.; Gao, X. Rhizoma Amorphophalli inhibits TNBC cell proliferation, migration, invasion and metastasis through the PI3K/Akt/mTOR pathway. J. Ethnopharmacol. 2018, 211, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Jiang, P.; Liu, W.; Xu, H.; Li, Y.; Li, Z.; Ma, H.; Yu, Y.; Li, X.; Ye, X. Role of Daucosterol Linoleate on Breast Cancer: Studies on Apoptosis and Metastasis. J. Agric. Food Chem. 2018, 66, 6031–6041. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Giampieri, F.; Gasparrini, M.; Forbes-Hernández, T.Y.; Cianciosi, D.; Reboredo-Rodriguez, P.; Manna, P.P.; Zhang, J.; Quiles, J.L.; Battino, M. The inhibitory effect of Manuka honey on human colon cancer HCT-116 and LoVo cell growth. Part 2: Induction of oxidative stress, alteration of mitochondrial respiration and glycolysis, and suppression of metastatic ability. Food Funct. 2018, 9, 2158–2170. [Google Scholar] [CrossRef] [PubMed]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.-Y. ROS-Mediated Cancer Cell Killing through Dietary Phytochemicals. Oxid. Med. Cell. Longev. 2019, 2019, 9051542. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, A. Chemoprevention with phytochemicals targeting inducible nitric oxide synthase. Forum Nutr. 2009, 61, 193–203. [Google Scholar] [CrossRef]
- Tedeschi, E.; Menegazzi, M.; Margotto, D.; Suzuki, H.; Förstermann, U.; Kleinert, H. Anti-inflammatory actions of St. John’s wort: Inhibition of human inducible nitric-oxide synthase expression by down-regulating signal transducer and activator of transcription-1alpha (STAT-1alpha) activation. J. Pharm. Exp. 2003, 307, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Menegazzi, M.; Novelli, M.; Beffy, P.; D’Aleo, V.; Tedeschi, E.; Lupi, R.; Zoratti, E.; Marchetti, P.; Suzuki, H.; Masiello, P. Protective effects of St. John’s wort extract and its component hyperforin against cytokine-induced cytotoxicity in a pancreatic beta-cell line. Int. J. Biochem. Cell Biol. 2008, 40, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef]
- Landry, W.D.; Cotter, T.G. ROS signalling, NADPH oxidases and cancer. Biochem. Soc. Trans. 2014, 42, 934–938. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef] [Green Version]
- Renard, P.; Zachary, M.D.; Bougelet, C.; Mirault, M.E.; Haegeman, G.; Remacle, J.; Raes, M. Effects of antioxidant enzyme modulations on interleukin-1-induced nuclear factor kappa B activation. Biochem. Pharm. 1997, 53, 149–160. [Google Scholar] [CrossRef]
- Fouani, L.; Kovacevic, Z.; Richardson, D.R. Targeting Oncogenic Nuclear Factor Kappa B Signaling with Redox-Active Agents for Cancer Treatment. Antioxid. Redox Signal. 2019, 30, 1096–1123. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharm. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Lim, J.K.M.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, K.; Hrabec, E.; Fichna, J.; Sosnowska, D.; Koziołkiewicz, M.; Szymański, J.; Lewandowska, U. Flavanols from Japanese quince (Chaenomeles japonica) fruit suppress expression of cyclooxygenase-2, metalloproteinase-9, and nuclear factor-kappaB in human colon cancer cells. Acta Biochim. Pol. 2017, 64, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Moskaug, J.Ø.; Carlsen, H.; Myhrstad, M.C.W.; Blomhoff, R. Polyphenols and glutathione synthesis regulation. Am. J. Clin. Nutr. 2005, 81, 277S–283S. [Google Scholar] [CrossRef] [PubMed]
- Stefanson, A.L.; Bakovic, M. Dietary regulation of Keap1/Nrf2/ARE pathway: Focus on plant-derived compounds and trace minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval-Acuña, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef]
- León-González, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-oxidant activity of polyphenols and its implication on cancer chemoprevention and chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380. [Google Scholar] [CrossRef]
- Hadi, S.; Bhat, S.; Azmi, A.; Hanif, S.; Shamim, U.; Ullah, M. Oxidative breakage of cellular DNA by plant polyphenols: A putative mechanism for anticancer properties. Semin. Cancer Biol. 2007, 17, 370–376. [Google Scholar] [CrossRef]
- Nakazato, T.; Ito, K.; Miyakawa, Y.; Kinjo, K.; Yamada, T.; Hozumi, N.; Ikeda, Y.; Kizaki, M. Catechin, a green tea component, rapidly induces apoptosis of myeloid leukemic cells via modulation of reactive oxygen species production in vitro and inhibits tumor growth in vivo. Haematologica 2005, 90, 317–325. [Google Scholar]
- Jeong, J.C.; Jang, S.W.; Kim, T.H.; Kwon, C.H.; Kim, Y.K. Mulberry Fruit (Moris fructus) Extracts Induce Human Glioma Cell Death In Vitro Through ROS-Dependent Mitochondrial Pathway and Inhibits Glioma Tumor Growth In Vivo. Nutr. Cancer 2010, 62, 402–412. [Google Scholar] [CrossRef]
- Gundala, S.R.; Yang, C.; Mukkavilli, R.; Paranjpe, R.; Brahmbhatt, M.; Pannu, V.; Cheng, A.; Reid, M.D.; Aneja, R. Hydroxychavicol, a betel leaf component, inhibits prostate cancer through ROS-driven DNA damage and apoptosis. Toxicol. Appl. Pharmacol. 2014, 280, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-X.; Chen, Y.-K.; Hou, Z.; Xiao, H.; Jin, H.; Lu, G.; Lee, M.-J.; Liu, B.; Guan, F.; Yang, Z.; et al. Pro-oxidative activities and dose-response relationship of (-)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: A comparative study in vivo and in vitro. Carcinogenesis 2010, 31, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys. 2008, 476, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Leslie, T.K.; James, A.D.; Zaccagna, F.; Grist, J.T.; Deen, S.; Kennerley, A.; Riemer, F.; Kaggie, J.D.; Gallagher, F.A.; Gilbert, F.J.; et al. Sodium homeostasis in the tumour microenvironment. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188304. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Alfarouk, K.O.; Elliott, R.L.; Alqahtani, S.S.; Ahmed, S.B.M.; Aljarbou, A.N.; Greco, M.R.; Cannone, S.; Reshkin, S.J. The Role of Sodium Hydrogen Exchanger 1 in Dysregulation of Proton Dynamics and Reprogramming of Cancer Metabolism as a Sequela. Int. J. Mol. Sci. 2019, 20, 3694. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Fong, S.; Baksh, S.; Fliegel, L. Na (+)/H (+)exchange in the tumour microenvironment: Does NHE1 drive breast cancer carcinogenesis? Int. J. Dev. Biol. 2015, 59, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Fliegel, L. Na+/H+ exchanger-mediated hydrogen ion extrusion as a carcinogenic signal in triple-negative breast cancer etiopathogenesis and prospects for its inhibition in therapeutics. Semin. Cancer Biol. 2017, 43, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na+/H+ exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective? Semin. Cancer Biol. 2017, 43, 5–16. [Google Scholar] [CrossRef]
- McIntyre, A.; Hulikova, A.; Ledaki, I.; Snell, C.; Singleton, D.; Steers, G.; Seden, P.; Jones, D.; Bridges, E.; Wigfield, S.; et al. Disrupting Hypoxia-Induced Bicarbonate Transport Acidifies Tumor Cells and Suppresses Tumor Growth. Cancer Res. 2016, 76, 3744–3755. [Google Scholar] [CrossRef] [Green Version]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Daniel, C.; Bell, C.; Burton, C.; Harguindey, S.; Reshkin, S.J.; Rauch, C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim. Biophys. Acta 2013, 1832, 606–617. [Google Scholar] [CrossRef]
- Harguindey, S.; Orive, G.; Luis Pedraz, J.; Paradiso, A.; Reshkin, S.J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin--one single nature. Biochim. Biophys. Acta 2005, 1756, 1–24. [Google Scholar] [CrossRef]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Reed, J.C. Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ. 2000, 7, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tsauo, J.; Geng, C.; Zhao, H.; Lei, X.; Li, X. Ginsenoside Rg3 Decreases NHE1 Expression via Inhibiting EGF-EGFR-ERK1/2-HIF-1 α Pathway in Hepatocellular Carcinoma: A Novel Antitumor Mechanism. Am. J. Chin. Med. 2018, 46, 1915–1931. [Google Scholar] [CrossRef]
- Abdelazeem, K.N.M.; Singh, Y.; Lang, F.; Salker, M.S. Negative Effect of Ellagic Acid on Cytosolic pH Regulation and Glycolytic Flux in Human Endometrial Cancer Cells. Cell. Physiol. Biochem. 2017, 41, 2374–2382. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.; Sim, S.; Park, S.H.; Song, K.D.; Lee, H.-K.; Heo, T.-H.; Jun, H.S.; Kim, S.-J. Anti-proliferative effect of Zea mays L. cob extract on rat C6 glioma cells through regulation of glycolysis, mitochondrial ROS, and apoptosis. Biomed. Pharm. 2018, 98, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Terada, H. Uncouplers of oxidative phosphorylation. Environ. Health Perspect. 1990, 87, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Romaschenko, V.P.; Zinovkin, R.A.; Galkin, I.I.; Zakharova, V.V.; Panteleeva, A.A.; Tokarchuk, A.V.; Lyamzaev, K.G.; Pletjushkina, O.Y.; Chernyak, B.V.; Popova, E.N. Low Concentrations of Uncouplers of Oxidative Phosphorylation Prevent Inflammatory Activation of Endothelial Cells by Tumor Necrosis Factor. Biochem. Mosc. 2015, 80, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.H.Z.; Pardo-Andreu, G.L.; Nuñez-Figueredo, Y.; Cuesta-Rubio, O.; Marín-Prida, J.; Uyemura, S.A.; Curti, C.; Alberici, L.C. Clusianone, a naturally occurring nemorosone regioisomer, uncouples rat liver mitochondria and induces HepG2 cell death. Chem. Biol. Interact. 2014, 212, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Li, J.; Mahdi, F.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.-D. Glycolysis inhibitor screening identifies the bis-geranylacylphloroglucinol protonophore moronone from Moronobea coccinea. J. Nat. Prod. 2012, 75, 2216–2222. [Google Scholar] [CrossRef] [Green Version]
- Sell, T.S.; Belkacemi, T.; Flockerzi, V.; Beck, A. Protonophore properties of hyperforin are essential for its pharmacological activity. Sci. Rep. 2014, 4, 7500. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.; Anderson, L.A.; Phillipson, J.D. St John’s wort (Hypericum perforatum L.): A review of its chemistry, pharmacology and clinical properties. J. Pharm. Pharm. 2001, 53, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Zirak, N.; Shafiee, M.; Soltani, G.; Mirzaei, M.; Sahebkar, A. Hypericum perforatum in the treatment of psychiatric and neurodegenerative disorders: Current evidence and potential mechanisms of action. J. Cell. Physiol. 2019, 234, 8496–8508. [Google Scholar] [CrossRef]
- Singer, A.; Wonnemann, M.; Müller, W.E. Hyperforin, a major antidepressant constituent of St. John’s Wort, inhibits serotonin uptake by elevating free intracellular Na+1. J. Pharm. Exp. 1999, 290, 1363–1368. [Google Scholar]
- Ng, Q.X.; Venkatanarayanan, N.; Ho, C.Y.X. Clinical use of Hypericum perforatum (St John’s wort) in depression: A meta-analysis. J. Affect. Disord. 2017, 210, 211–221. [Google Scholar] [CrossRef]
- Apaydin, E.A.; Maher, A.R.; Shanman, R.; Booth, M.S.; Miles, J.N.V.; Sorbero, M.E.; Hempel, S. A systematic review of St. John’s wort for major depressive disorder. Syst. Rev. 2016, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Siracusa, L.; Ruberto, G.; Carrubba, A.; Lazzara, S.; Speciale, A.; Cimino, F.; Saija, A.; Cristani, M. Phytochemical profiles, phototoxic and antioxidant properties of eleven Hypericum species—A comparative study. Phytochemistry 2018, 152, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; Sacchetti, G. Factors affecting polyphenol biosynthesis in wild and field grown St. John’s Wort (Hypericum perforatum L. Hypericaceae/Guttiferae). Molecules 2009, 14, 682–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyis, F.; Yurteri, E.; Özcan, A.; Cirak, C. Altitudinal impacts on chemical content and composition of Hypericum perforatum, a prominent medicinal herb. S. Afr. J. Bot. 2020, 135, 391–403. [Google Scholar] [CrossRef]
- Krammer, B.; Verwanger, T. Molecular response to hypericin-induced photodamage. Curr. Med. Chem. 2012, 19, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yin, G.; Le, V.; Zhang, A.; Lu, Y.; Yang, M.; Fei, Z.; Liu, J. Hypericin-based Photodynamic Therapy Induces a Tumor-Specific Immune Response and an Effective DC-based cancer Immunotherapy. Biochem. Pharm. 2014. [Google Scholar] [CrossRef] [PubMed]
- Albert, D.; Zündorf, I.; Dingermann, T.; Müller, W.E.; Steinhilber, D.; Werz, O. Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem. Pharm. 2002, 64, 1767–1775. [Google Scholar] [CrossRef]
- Feisst, C.; Pergola, C.; Rakonjac, M.; Rossi, A.; Koeberle, A.; Dodt, G.; Hoffmann, M.; Hoernig, C.; Fischer, L.; Steinhilber, D.; et al. Hyperforin is a novel type of 5-lipoxygenase inhibitor with high efficacy in vivo. Cell. Mol. Life Sci. 2009, 66, 2759–2771. [Google Scholar] [CrossRef] [PubMed]
- Novelli, M.; Menegazzi, M.; Beffy, P.; Porozov, S.; Gregorelli, A.; Giacopelli, D.; De Tata, V.; Masiello, P. St. John’s wort extract and hyperforin inhibit multiple phosphorylation steps of cytokine signaling and prevent inflammatory and apoptotic gene induction in pancreatic β cells. Int. J. Biochem. Cell Biol. 2016, 81, 92–104. [Google Scholar] [CrossRef]
- Biber, A.; Fischer, H.; Römer, A.; Chatterjee, S.S. Oral bioavailability of hyperforin from hypericum extracts in rats and human volunteers. Pharmacopsychiatry 1998, 31 (Suppl. 1), 36–43. [Google Scholar] [CrossRef]
- Schulz, H.-U.; Schürer, M.; Bässler, D.; Weiser, D. Investigation of the bioavailability of hypericin, pseudohypericin, hyperforin and the flavonoids quercetin and isorhamnetin following single and multiple oral dosing of a hypericum extract containing tablet. Arzneimittelforschung 2005, 55, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Kraus, B.; Wolff, H.; Elstner, E.F.; Heilmann, J. Hyperforin is a modulator of inducible nitric oxide synthase and phagocytosis in microglia and macrophages. Naunyn. Schmiedebergs Arch. Pharm. 2010, 381, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Novelli, M.; Beffy, P.; Gregorelli, A.; Porozov, S.; Mascia, F.; Vantaggiato, C.; Masiello, P.; Menegazzi, M. Persistence of STAT-1 inhibition and induction of cytokine resistance in pancreatic β cells treated with St John’s wort and its component hyperforin. J. Pharm. Pharm. 2019, 71, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, H.C.; Rentel, C.; Schmidt, P.C. Isolation, purity analysis and stability of hyperforin as a standard material from Hypericum perforatum L. J. Pharm. Pharm. 1999, 51, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, M.; Moia, M.; Funicello, M.; Riva, A.; Morazzoni, P.; Mennini, T. In vitro effects of the dicyclohexylammonium salt of hyperforin on interleukin-6 release in different experimental models. Planta Med. 2004, 70, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Donà, M.; Dell’Aica, I.; Pezzato, E.; Sartor, L.; Calabrese, F.; Della Barbera, M.; Donella-Deana, A.; Appendino, G.; Borsarini, A.; Caniato, R.; et al. Hyperforin inhibits cancer invasion and metastasis. Cancer Res. 2004, 64, 6225–6232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartner, M.; Müller, T.; Simon, J.C.; Giannis, A.; Sleeman, J.P. Aristoforin, a novel stable derivative of hyperforin, is a potent anticancer agent. Chembiochem 2005, 6, 171–177. [Google Scholar] [CrossRef]
- Hammer, K.D.P.; Hillwig, M.L.; Solco, A.K.S.; Dixon, P.M.; Delate, K.; Murphy, P.A.; Wurtele, E.S.; Birt, D.F. Inhibition of prostaglandin E(2) production by anti-inflammatory hypericum perforatum extracts and constituents in RAW264.7 Mouse Macrophage Cells. J. Agric. Food Chem. 2007, 55, 7323–7331. [Google Scholar] [CrossRef] [Green Version]
- Koeberle, A.; Rossi, A.; Bauer, J.; Dehm, F.; Verotta, L.; Northoff, H.; Sautebin, L.; Werz, O. Hyperforin, an Anti-Inflammatory Constituent from St. John’s Wort, Inhibits Microsomal Prostaglandin E(2) Synthase-1 and Suppresses Prostaglandin E(2) Formation in vivo. Front. Pharm. 2011, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Novelli, M.; Beffy, P.; Menegazzi, M.; De Tata, V.; Martino, L.; Sgarbossa, A.; Porozov, S.; Pippa, A.; Masini, M.; Marchetti, P.; et al. St. John’s wort extract and hyperforin protect rat and human pancreatic islets against cytokine toxicity. Acta Diabetol. 2014, 51, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Novelli, M.; Masiello, P.; Beffy, P.; Menegazzi, M. Protective Role of St. John’s Wort and Its Components Hyperforin and Hypericin against Diabetes through Inhibition of Inflammatory Signaling: Evidence from In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 8108. [Google Scholar] [CrossRef]
- Müller, M.; Essin, K.; Hill, K.; Beschmann, H.; Rubant, S.; Schempp, C.M.; Gollasch, M.; Boehncke, W.H.; Harteneck, C.; Müller, W.E.; et al. Specific TRPC6 channel activation, a novel approach to stimulate keratinocyte differentiation. J. Biol. Chem. 2008, 283, 33942–33954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedí, J.; Arroyo, R.; Romero, C.; Martín-Aragón, S.; Villar, A.M. Antioxidant properties and protective effects of a standardized extract of Hypericum perforatum on hydrogen peroxide-induced oxidative damage in PC12 cells. Life Sci. 2004, 75, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Feisst, C.; Werz, O. Suppression of receptor-mediated Ca2+ mobilization and functional leukocyte responses by hyperforin. Biochem. Pharm. 2004, 67, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Kim, J.-E.; Kim, Y.-J.; Kim, M.-J.; Kang, T.-C. Hyperforin attenuates microglia activation and inhibits p65-Ser276 NFκB phosphorylation in the rat piriform cortex following status epilepticus. Neurosci. Res. 2014, 85, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shao, B.; Yu, H.; Xu, F.; Wang, P.; Yu, K.; Han, Y.; Song, M.; Li, Y.; Cao, Z. Neuroprotective role of hyperforin on aluminum maltolate-induced oxidative damage and apoptosis in PC12 cells and SH-SY5Y cells. Chem. Biol. Interact. 2019, 299, 15–26. [Google Scholar] [CrossRef]
- Menegazzi, M.; Di Paola, R.; Mazzon, E.; Muià, C.; Genovese, T.; Crisafulli, C.; Suzuki, H.; Cuzzocrea, S. Hypericum perforatum attenuates the development of carrageenan-induced lung injury in mice. Free Radic. Biol. Med. 2006, 40, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Mazzon, E.; Muià, C.; Crisafulli, C.; Genovese, T.; Di Bella, P.; Esposito, E.; Menegazzi, M.; Meli, R.; Suzuki, H.; et al. Protective effect of Hypericum perforatum in zymosan-induced multiple organ dysfunction syndrome: Relationship to its inhibitory effect on nitric oxide production and its peroxynitrite scavenging activity. Nitric Oxide 2007, 16, 118–130. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Di Paola, R.; Muià, C.; Crisafulli, C.; Menegazzi, M.; Malleo, G.; Suzuki, H.; Cuzzocrea, S. Hypericum perforatum attenuates the development of cerulein-induced acute pancreatitis in mice. Shock 2006, 25, 161–167. [Google Scholar] [CrossRef]
- De Paola, R.; Muià, C.; Mazzon, E.; Genovese, T.; Crisafulli, C.; Menegazzi, M.; Caputi, A.P.; Suzuki, H.; Cuzzocrea, S. Effects of Hypericum perforatum extract in a rat model of ischemia and reperfusion injury. Shock 2005, 24, 255–263. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Menegazzi, M.; Di Paola, R.; Muià, C.; Crisafulli, C.; Bramanti, P.; Suzuki, H.; Cuzzocrea, S. Neuroprotection and enhanced recovery with hypericum perforatum extract after experimental spinal cord injury in mice. Shock 2006, 25, 608–617. [Google Scholar] [CrossRef]
- Raso, G.M.; Pacilio, M.; Di Carlo, G.; Esposito, E.; Pinto, L.; Meli, R. In-vivo and in-vitro anti-inflammatory effect of Echinacea purpurea and Hypericum perforatum. J. Pharm. Pharm. 2002, 54, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Savikin, K.; Dobrić, S.; Tadić, V.; Zdunić, G. Antiinflammatory activity of ethanol extracts of Hypericum perforatum L., H. barbatum Jacq., H. hirsutum L., H. richeri Vill. and H. androsaemum L. in rats. Phytother. Res. 2007, 21, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.E. Anti-inflammatory, antinociceptive, and gastric effects of Hypericum perforatum in rats. Sci. World J. 2005, 5, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zdunić, G.; Godevac, D.; Milenković, M.; Vucićević, D.; Savikin, K.; Menković, N.; Petrović, S. Evaluation of Hypericum perforatum oil extracts for an antiinflammatory and gastroprotective activity in rats. Phytother. Res. 2009, 23, 1559–1564. [Google Scholar] [CrossRef]
- Dost, T.; Ozkayran, H.; Gokalp, F.; Yenisey, C.; Birincioglu, M. The effect of Hypericum perforatum (St. John’s Wort) on experimental colitis in rat. Dig. Dis. Sci. 2009, 54, 1214–1221. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Yang, X.-X.; Chan, S.Y.; Xu, A.-L.; Duan, W.; Zhu, Y.-Z.; Sheu, F.-S.; Boelsterli, U.A.; Chan, E.; Zhang, Q.; et al. St. John’s wort attenuates irinotecan-induced diarrhea via down-regulation of intestinal pro-inflammatory cytokines and inhibition of intestinal epithelial apoptosis. Toxicol. Appl. Pharm. 2006, 216, 225–237. [Google Scholar] [CrossRef]
- Hadzhiiliev, V.; Dimov, D. Separate isolation of hyperforin from Hypericum perforatum (St. John`s Wort) pursuant to the coefficents LOG Kow, PKa and densities of the included compounds. TJS 2015, 13, 19–23. [Google Scholar] [CrossRef]
- Roz, N.; Rehavi, M. Hyperforin inhibits vesicular uptake of monoamines by dissipating pH gradient across synaptic vesicle membrane. Life Sci. 2003, 73, 461–470. [Google Scholar] [CrossRef]
- Froestl, B.; Steiner, B.; Müller, W.E. Enhancement of proteolytic processing of the beta-amyloid precursor protein by hyperforin. Biochem. Pharm. 2003, 66, 2177–2184. [Google Scholar] [CrossRef]
- Friedland, K.; Harteneck, C. Hyperforin: To Be or Not to Be an Activator of TRPC(6). In Reviews of Physiology, Biochemistry and Pharmacology Vol. 169; Nilius, B., Gudermann, T., Jahn, R., Lill, R., Petersen, O.H., de Tombe, P.P., Eds.; Springer: Cham, Switzerland, 2015; Volume 169, pp. 1–24. ISBN 978-3-319-26563-6. [Google Scholar]
- Tu, P.; Gibon, J.; Bouron, A. The TRPC6 channel activator hyperforin induces the release of zinc and calcium from mitochondria. J. Neurochem. 2010, 112, 204–213. [Google Scholar] [CrossRef]
- Werz, O.; Steinhilber, D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Ther. 2006, 112, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Imreova, P.; Feruszova, J.; Kyzek, S.; Bodnarova, K.; Zduriencikova, M.; Kozics, K.; Mucaji, P.; Galova, E.; Sevcovicova, A.; Miadokova, E.; et al. Hyperforin Exhibits Antigenotoxic Activity on Human and Bacterial Cells. Molecules 2017, 22, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinke, M.C.; Schanzer, S.; Haag, S.F.; Casetti, F.; Müller, M.L.; Wölfle, U.; Kleemann, A.; Lademann, J.; Schempp, C.M. In vivo photoprotective and anti-inflammatory effect of hyperforin is associated with high antioxidant activity in vitro and ex vivo. Eur. J. Pharm. Biopharm. 2012, 81, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Ševčovičová, A.; Šemeláková, M.; Plšíková, J.; Loderer, D.; Imreová, P.; Gálová, E.; Kožurková, M.; Miadoková, E.; Fedoročko, P. DNA-protective activities of hyperforin and aristoforin. Toxicol. In Vitro 2015, 29, 631–637. [Google Scholar] [CrossRef]
- Hecht, S.M. Bleomycin: New perspectives on the mechanism of action. J. Nat. Prod. 2000, 63, 158–168. [Google Scholar] [CrossRef]
- Kurze, A.-K.; Buhs, S.; Eggert, D.; Oliveira-Ferrer, L.; Müller, V.; Niendorf, A.; Wagener, C.; Nollau, P. Immature O-glycans recognized by the macrophage glycoreceptor CLEC10A (MGL) are induced by 4-hydroxy-tamoxifen, oxidative stress and DNA-damage in breast cancer cells. Cell Commun. Signal. 2019, 17, 107. [Google Scholar] [CrossRef] [Green Version]
- Lagadic-Gossmann, D.; Hardonnière, K.; Mograbi, B.; Sergent, O.; Huc, L. Disturbances in H+ dynamics during environmental carcinogenesis. Biochimie 2019, 163, 171–183. [Google Scholar] [CrossRef]
- Manna, S.K.; Golla, S.; Golla, J.P.; Tanaka, N.; Cai, Y.; Takahashi, S.; Krausz, K.W.; Matsubara, T.; Korboukh, I.; Gonzalez, F.J. St. John’s Wort Attenuates Colorectal Carcinogenesis in Mice through Suppression of Inflammatory Signaling. Cancer Prev. Res. 2015, 8, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Kuai, Y.; Liu, H.; Liu, D.; Liu, Y.; Sun, Y.; Xie, J.; Sun, J.; Fang, Y.; Pan, H.; Han, W. An ultralow dose of the NADPH oxidase inhibitor diphenyleneiodonium (DPI) is an economical and effective therapeutic agent for the treatment of colitis-associated colorectal cancer. Theranostics 2020, 10, 6743–6757. [Google Scholar] [CrossRef]
- Rahimi, R.; Abdollahi, M. An update on the ability of St. John’s wort to affect the metabolism of other drugs. Expert Opin. Drug Metab. Toxicol. 2012, 8, 691–708. [Google Scholar] [CrossRef]
- He, X.; Feng, S. Role of Metabolic Enzymes P450 (CYP) on Activating Procarcinogen and their Polymorphisms on the Risk of Cancers. Curr. Drug Metab. 2015, 16, 850–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.; Arlt, V.M.; Phillips, D.H. The role of cytochrome P450 enzymes in carcinogen activation and detoxication: An in vivo–in vitro paradox. Carcinogenesis 2018, 39, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Beyerle, J.; Holowatyj, A.N.; Haffa, M.; Frei, E.; Gigic, B.; Schrotz-King, P.; Boehm, J.; Habermann, N.; Stiborova, M.; Scherer, D.; et al. Expression Patterns of Xenobiotic-Metabolizing Enzymes in Tumor and Adjacent Normal Mucosa Tissues among Patients with Colorectal Cancer: The ColoCare Study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Satia, J.A.; Littman, A.; Slatore, C.G.; Galanko, J.A.; White, E. Associations of herbal and specialty supplements with lung and colorectal cancer risk in the VITamins and Lifestyle study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichling, J.; Weseler, A.; Saller, R. A Current Review of the Antimicrobial Activity of Hypericum perforatum L. Pharmacopsychiatry 2001, 34, 116–118. [Google Scholar] [CrossRef]
- Boyanova, L. Comparative evaluation of the activity of plant infusions against Helicobacter pylori strains by three methods. World J. Microbiol. Biotechnol. 2014, 30, 1633–1637. [Google Scholar] [CrossRef]
- Pang, R.; Tao, J.; Zhang, S.; Zhu, J.; Yue, X.; Zhao, L.; Ye, P.; Zhu, Y. In vitro anti-hepatitis B virus effect of Hypericum perforatum L. J. Huazhong Univ. Sci. Technol. 2010, 30, 98–102. [Google Scholar] [CrossRef]
- Schempp, C.M.; Kirkin, V.; Simon-Haarhaus, B.; Kersten, A.; Kiss, J.; Termeer, C.C.; Gilb, B.; Kaufmann, T.; Borner, C.; Sleeman, J.P.; et al. Inhibition of tumour cell growth by hyperforin, a novel anticancer drug from St. John’s wort that acts by induction of apoptosis. Oncogene 2002, 21, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.-T.; Chen, W.-T.; Wu, C.-T.; Chung, J.-G. Hyperforin induces apoptosis through extrinsic/intrinsic pathways and inhibits EGFR/ERK/NF-κB-mediated anti-apoptotic potential in glioblastoma. Environ. Toxicol. 2020, 35, 1058–1069. [Google Scholar] [CrossRef]
- Wiechmann, K.; Müller, H.; Fischer, D.; Jauch, J.; Werz, O. The acylphloroglucinols hyperforin and myrtucommulone A cause mitochondrial dysfunctions in leukemic cells by direct interference with mitochondria. Apoptosis 2015, 20, 1508–1517. [Google Scholar] [CrossRef]
- Chiang, I.-T.; Chen, W.-T.; Tseng, C.-W.; Chen, Y.-C.; Kuo, Y.-C.; Chen, B.-J.; Weng, M.-C.; Lin, H.-J.; Wang, W.-S. Hyperforin Inhibits Cell Growth by Inducing Intrinsic and Extrinsic Apoptotic Pathways in Hepatocellular Carcinoma Cells. Anticancer Res. 2017, 37, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naven, R.T.; Swiss, R.; Klug-McLeod, J.; Will, Y.; Greene, N. The development of structure-activity relationships for mitochondrial dysfunction: Uncoupling of oxidative phosphorylation. Toxicol. Sci. 2013, 131, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo-Andreu, G.L.; Nuñez-Figueredo, Y.; Tudella, V.G.; Cuesta-Rubio, O.; Rodrigues, F.P.; Pestana, C.R.; Uyemura, S.A.; Leopoldino, A.M.; Alberici, L.C.; Curti, C. The anti-cancer agent nemorosone is a new potent protonophoric mitochondrial uncoupler. Mitochondrion 2011, 11, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Kim, S.W.; Kim, S.H.; Kim, S.Z.; Park, W.H. 2,4-dinitrophenol induces G1 phase arrest and apoptosis in human pulmonary adenocarcinoma Calu-6 cells. Toxicol. In Vitro 2008, 22, 659–670. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, A.O.; van den Heuvel, L.P.; Dijkman, H.B.P.M.; de Abreu, R.A.; Birkenkamp, K.U.; de Witte, T.; van der Reijden, B.A.; Smeitink, J.A.M.; Jansen, J.H. Bcl-2 prevents loss of mitochondria in CCCP-induced apoptosis. Exp. Cell Res. 2004, 299, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.A.; Seol, J.-W.; Kang, S.-J.; Park, S.-Y. Mitochondrial transmembrane potential and reactive oxygen species generation regulate the enhanced effect of CCCP on TRAIL-induced SNU-638 cell apoptosis. J. Vet. Med. Sci. 2008, 70, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 968–977. [Google Scholar] [CrossRef]
- Alp, H.; Tutun, H.; Kaplan, H.; ŞïNgïRïK, E.; Altintaş, L. Meme Kanseri Hücre Hattında Hypericum perforatum Ekstresinin Apoptotik Etkilerinin Araştırılması. Harran Üniversitesi Vet. Fakültesi Derg. 2019, 8, 198–202. [Google Scholar] [CrossRef]
- Orhan, I.E.; Kartal, M.; Gülpinar, A.R.; Yetkin, G.; Orlikova, B.; Diederich, M.; Tasdemir, D. Inhibitory effect of St. John׳s Wort oil macerates on TNFα-induced NF-κB activation and their fatty acid composition. J. Ethnopharmacol. 2014, 155, 1086–1092. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Lin, K.-H.; Hsieh, J.-H.; Chung, J.-G.; Tan, Z.-L.; Hsu, F.-T.; Chiang, C.-H. Hyperforin Induces Apoptosis Through Extrinsic/Intrinsic Pathways and Inhibits NF-ĸB-modulated Survival and Invasion Potential in Bladder Cancer. In Vivo 2019, 33, 1865–1877. [Google Scholar] [CrossRef]
- You, M.-K.; Kim, H.-J.; Kook, J.H.; Kim, H.-A. St. John’s Wort Regulates Proliferation and Apoptosis in MCF-7 Human Breast Cancer Cells by Inhibiting AMPK/mTOR and Activating the Mitochondrial Pathway. Int. J. Mol. Sci. 2018, 19, 966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merhi, F.; Tang, R.; Piedfer, M.; Mathieu, J.; Bombarda, I.; Zaher, M.; Kolb, J.-P.; Billard, C.; Bauvois, B. Hyperforin inhibits Akt1 kinase activity and promotes caspase-mediated apoptosis involving Bad and Noxa activation in human myeloid tumor cells. PLoS ONE 2011, 6, e25963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostanska, K.; Reichling, J.; Bommer, S.; Weber, M.; Saller, R. Hyperforin a constituent of St John’s wort (Hypericum perforatum L.) extract induces apoptosis by triggering activation of caspases and with hypericin synergistically exerts cytotoxicity towards human malignant cell lines. Eur. J. Pharm. Biopharm. 2003, 56, 121–132. [Google Scholar] [CrossRef]
- Quiney, C.; Billard, C.; Faussat, A.M.; Salanoubat, C.; Ensaf, A.; Naït-Si, Y.; Fourneron, J.D.; Kolb, J.-P. Pro-apoptotic properties of hyperforin in leukemic cells from patients with B-cell chronic lymphocytic leukemia. Leukemia 2006, 20, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Zaher, M.; Tang, R.; Bombarda, I.; Merhi, F.; Bauvois, B.; Billard, C. Hyperforin induces apoptosis of chronic lymphocytic leukemia cells through upregulation of the BH3-only protein Noxa. Int. J. Oncol. 2012, 40, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, L.A.; Hallaert, D.Y.H.; Spijker, R.; de Goeij, B.; Jaspers, A.; Kater, A.P.; van Oers, M.H.J.; van Noesel, C.J.M.; Eldering, E. Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocytic leukemia cells correlates with survival capacity. Blood 2007, 109, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Lim, H.Y.; Wong, K.P. Uncoupling of oxidative phosphorylation by curcumin: Implication of its cellular mechanism of action. Biochem. Biophys. Res. Commun. 2009, 389, 187–192. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kay, N.E.; Secreto, C.R.; Shanafelt, T.D. Curcumin inhibits prosurvival pathways in chronic lymphocytic leukemia B cells and may overcome their stromal protection in combination with EGCG. Clin. Cancer Res. 2009, 15, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieminen, A.I.; Eskelinen, V.M.; Haikala, H.M.; Tervonen, T.A.; Yan, Y.; Partanen, J.I.; Klefström, J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, E1839–E1848. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Poveda, B.; Verotta, L.; Bombardelli, E.; Quesada, A.R.; Medina, M.A. Tetrahydrohyperforin and octahydrohyperforin are two new potent inhibitors of angiogenesis. PLoS ONE 2010, 5, e9558. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Poveda, B.; Quesada, A.R.; Medina, M.A. Hyperforin, a bio-active compound of St. John’s Wort, is a new inhibitor of angiogenesis targeting several key steps of the process. Int. J. Cancer 2005, 117, 775–780. [Google Scholar] [CrossRef]
- Lorusso, G.; Vannini, N.; Sogno, I.; Generoso, L.; Garbisa, S.; Noonan, D.M.; Albini, A. Mechanisms of Hyperforin as an anti-angiogenic angioprevention agent. Eur. J. Cancer 2009, 45, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Rothley, M.; Schmid, A.; Thiele, W.; Schacht, V.; Plaumann, D.; Gartner, M.; Yektaoglu, A.; Bruyère, F.; Noël, A.; Giannis, A.; et al. Hyperforin and aristoforin inhibit lymphatic endothelial cell proliferation in vitro and suppress tumor-induced lymphangiogenesis in vivo. Int. J. Cancer 2009, 125, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Adjei, A.A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncology 2015, 20, 660–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A.; Carpenter, L.R.; Lakshminarayanan, V.; Page, S.M.; Moy, J.N.; Thomas, L.L. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J. Leukoc. Biol. 1999, 65, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [Green Version]
- Wyder, L.; Suply, T.; Ricoux, B.; Billy, E.; Schnell, C.; Baumgarten, B.U.; Maira, S.M.; Koelbing, C.; Ferretti, M.; Kinzel, B.; et al. Reduced pathological angiogenesis and tumor growth in mice lacking GPR4, a proton sensing receptor. Angiogenesis 2011, 14, 533–544. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef] [Green Version]
- Quiney, C.; Billard, C.; Mirshahi, P.; Fourneron, J.-D.; Kolb, J.-P. Hyperforin inhibits MMP-9 secretion by B-CLL cells and microtubule formation by endothelial cells. Leukemia 2006, 20, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.; Lee, Y.; Kim, C. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERKs and PI-3K/AKT pathways: Involvement of invasive potential. FASEB J. 2004, 18, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Karaarslan, S.; Cokmert, S.; Cokmez, A. Does St. John’s Wort cause regression in gastrointestinal system adenocarcinomas? World J. Gastrointest. Oncol. 2015, 7, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Heerdt, B.G.; Houston, M.A.; Augenlicht, L.H. The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res. 2005, 65, 9861–9867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandel, B.A.; Ekins, S.; Leuner, K.; Thasler, W.E.; Harteneck, C.; Zanger, U.M. No activation of human pregnane X receptor by hyperforin-related phloroglucinols. J. Pharm. Exp. 2014, 348, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menegazzi, M.; Masiello, P.; Novelli, M. Anti-Tumor Activity of Hypericum perforatum L. and Hyperforin through Modulation of Inflammatory Signaling, ROS Generation and Proton Dynamics. Antioxidants 2021, 10, 18. https://doi.org/10.3390/antiox10010018
Menegazzi M, Masiello P, Novelli M. Anti-Tumor Activity of Hypericum perforatum L. and Hyperforin through Modulation of Inflammatory Signaling, ROS Generation and Proton Dynamics. Antioxidants. 2021; 10(1):18. https://doi.org/10.3390/antiox10010018
Chicago/Turabian StyleMenegazzi, Marta, Pellegrino Masiello, and Michela Novelli. 2021. "Anti-Tumor Activity of Hypericum perforatum L. and Hyperforin through Modulation of Inflammatory Signaling, ROS Generation and Proton Dynamics" Antioxidants 10, no. 1: 18. https://doi.org/10.3390/antiox10010018
APA StyleMenegazzi, M., Masiello, P., & Novelli, M. (2021). Anti-Tumor Activity of Hypericum perforatum L. and Hyperforin through Modulation of Inflammatory Signaling, ROS Generation and Proton Dynamics. Antioxidants, 10(1), 18. https://doi.org/10.3390/antiox10010018