Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects, Legal Issues, Randomization, and Statistical Planning
2.2. Experimental Groups
2.3. Cerebral Ischemia/Reperfusion Model
2.4. Neurological Deficit Assessments
2.5. Assessment of Infarct Size by 2,3,5-Triphenyltetrazolium Chloride (TTC) Staining
2.6. Preparation of Brain Membranes
2.7. Screening CYP2E1 Enzyme Activity
2.8. Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Assay
2.9. Analysis of Inflammation
2.10. Enzyme-Linked Immunosorbent Essay Analysis (ELISA)
2.11. Analysis of Oxidative Stress
2.12. NeuroImaging
2.13. Statistical Analysis
3. Results
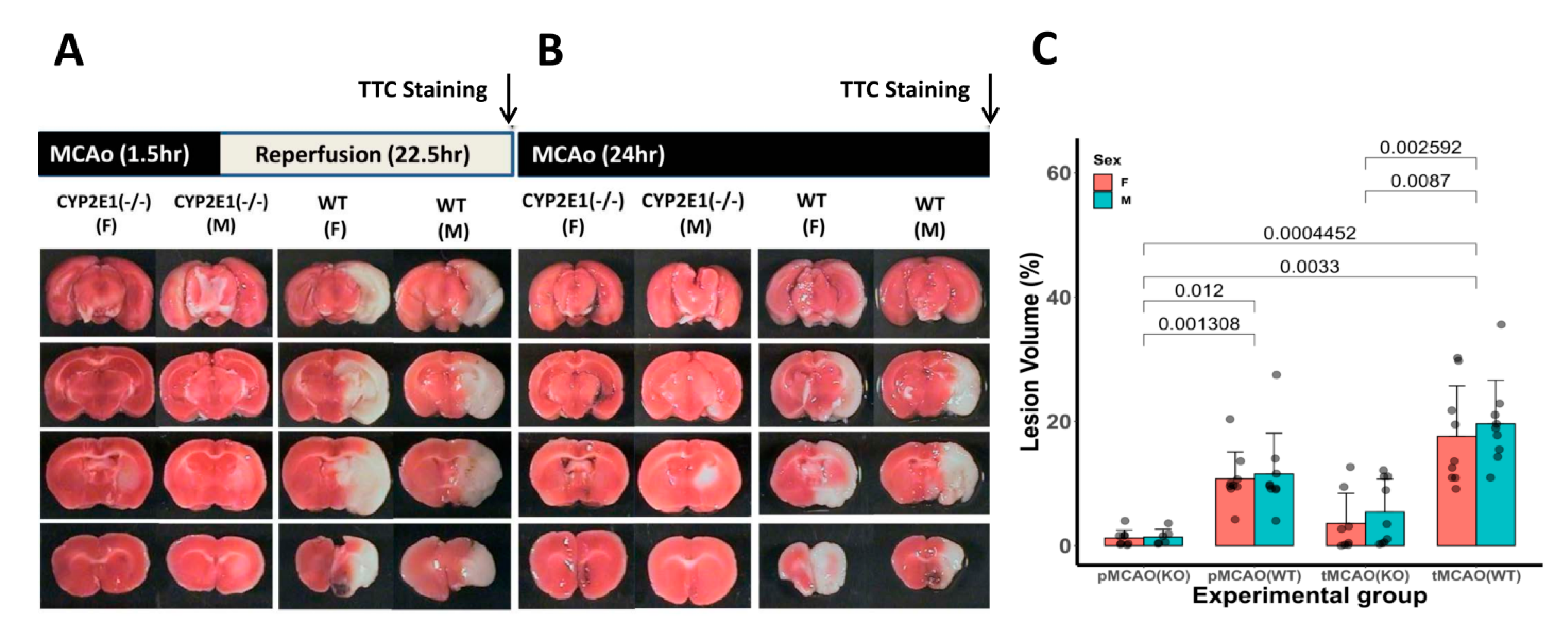
3.1. Infarct Volume and Neurological Outcomes
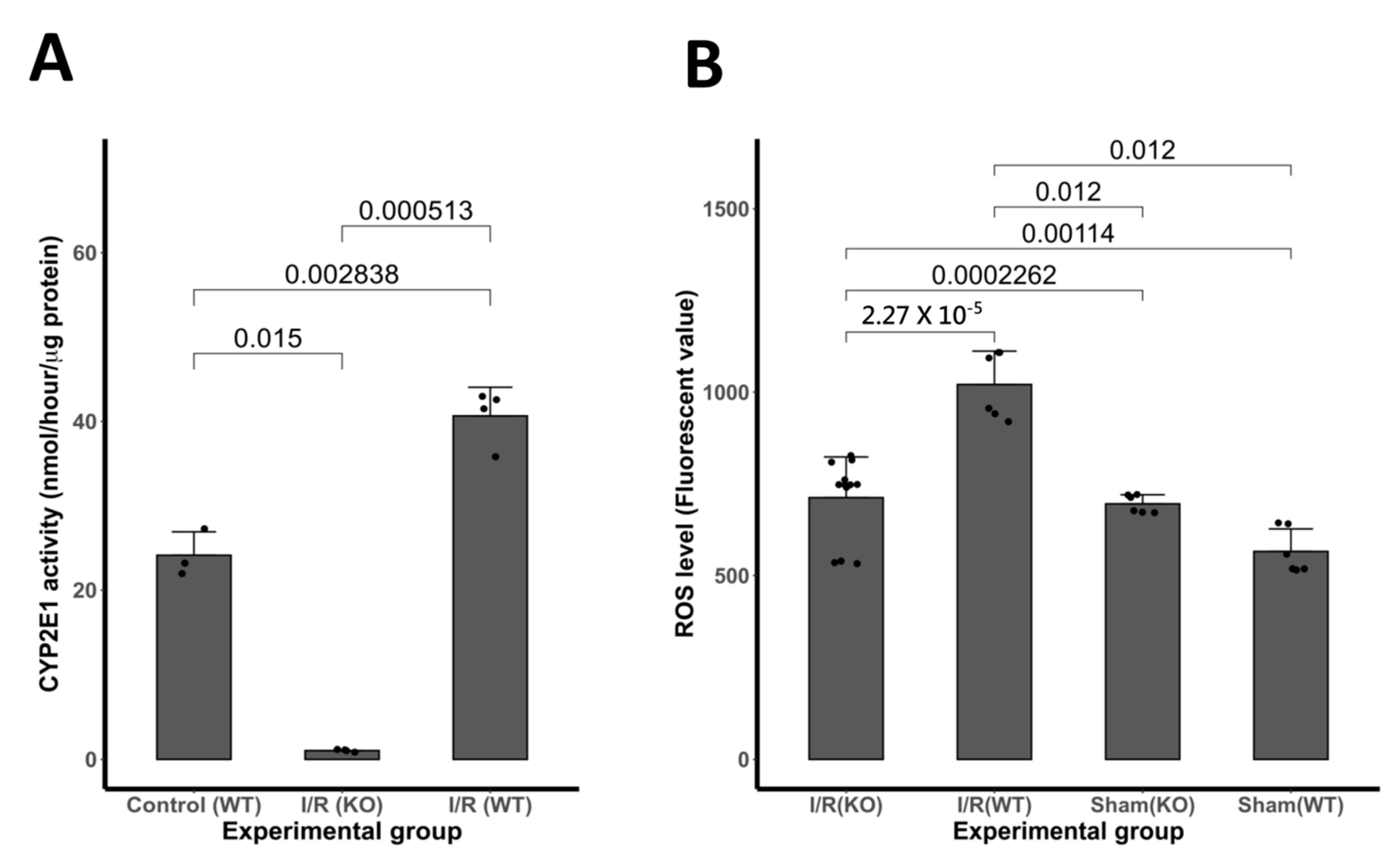
3.2. Expression of CYP2E1 Following I/R Insult
3.3. Expression of ROS Activity Following I/R Insult
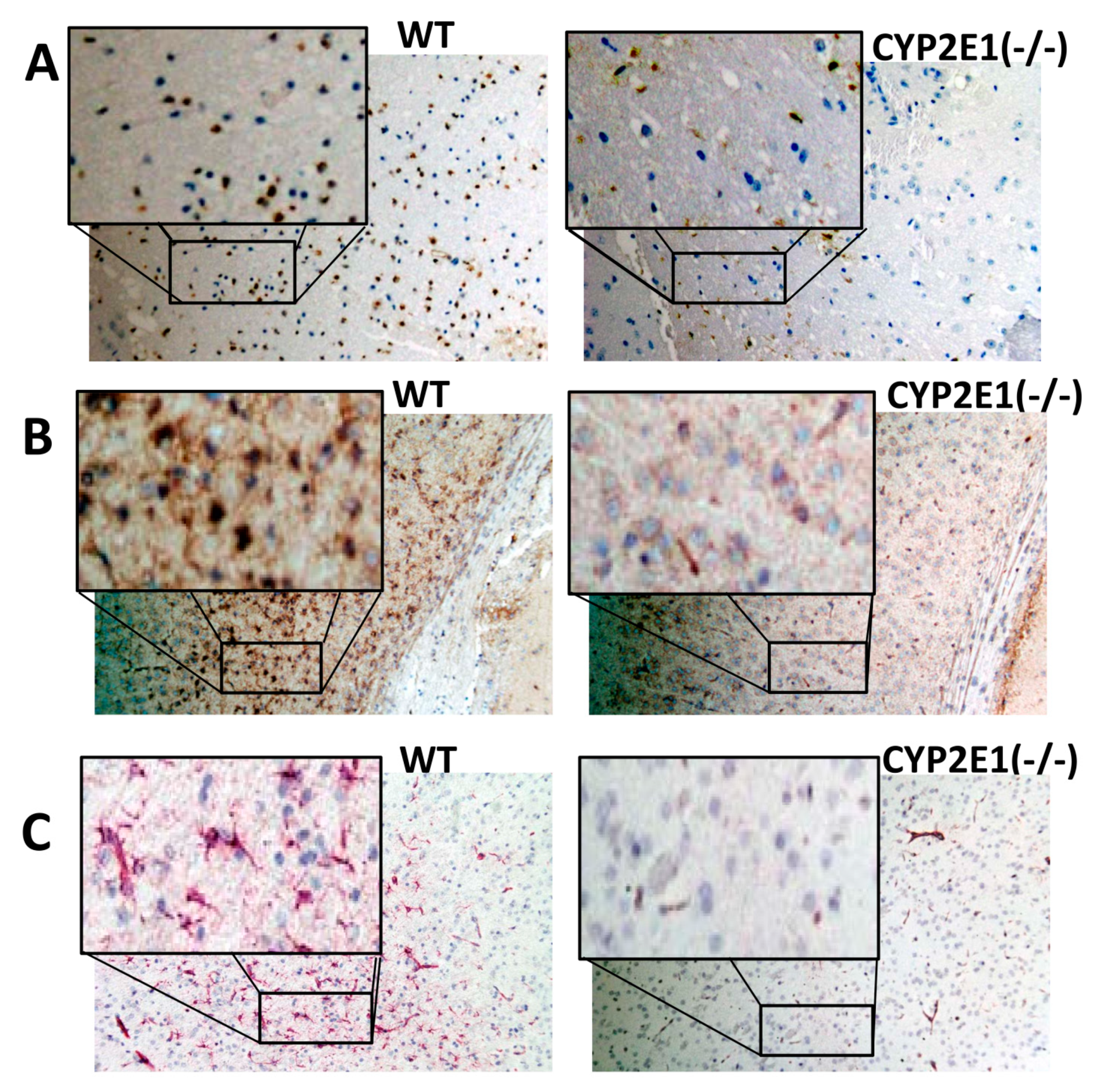
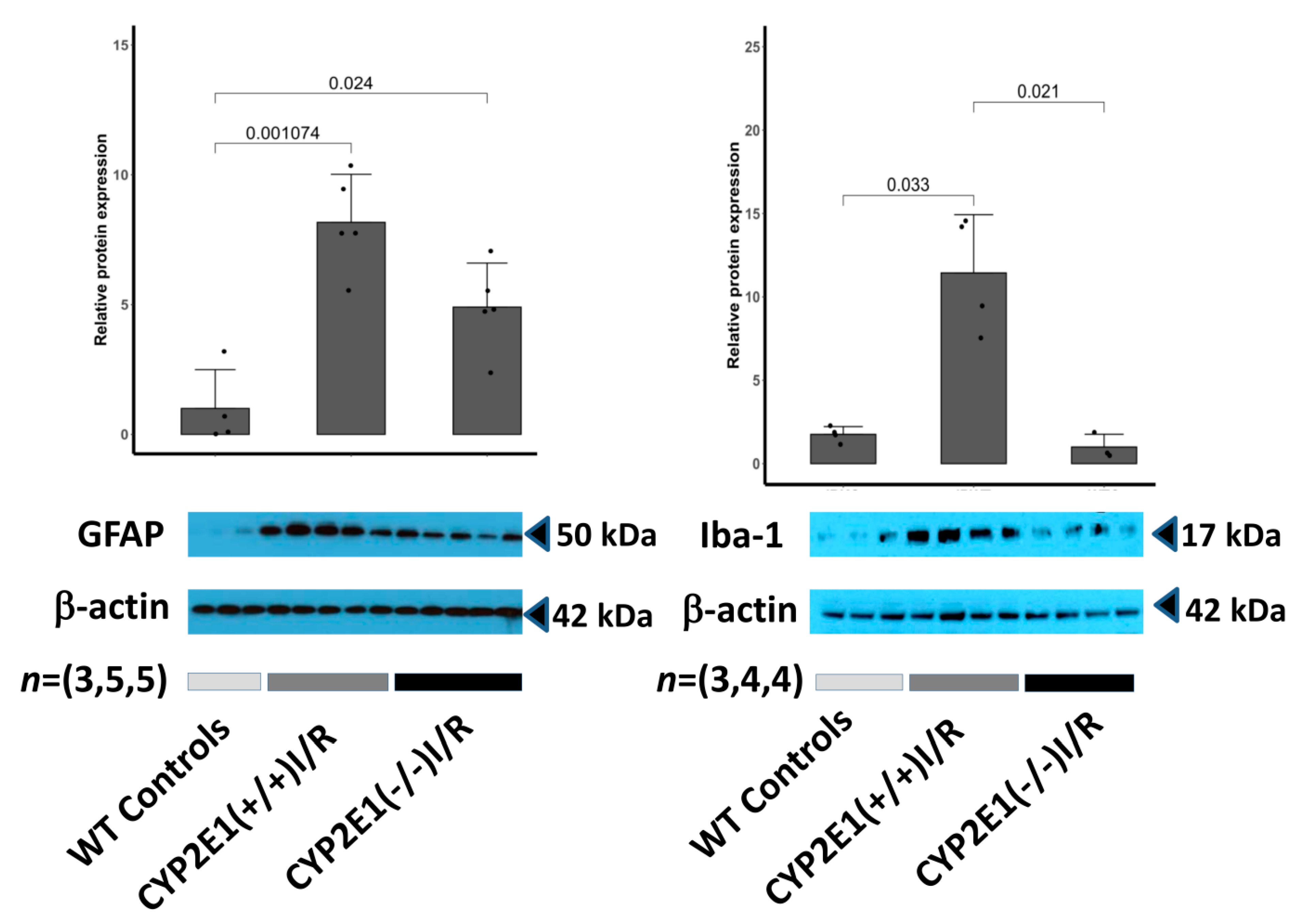
3.4. Cytoprotective Effects of CYP2E1 Inhibition and Glial Activation
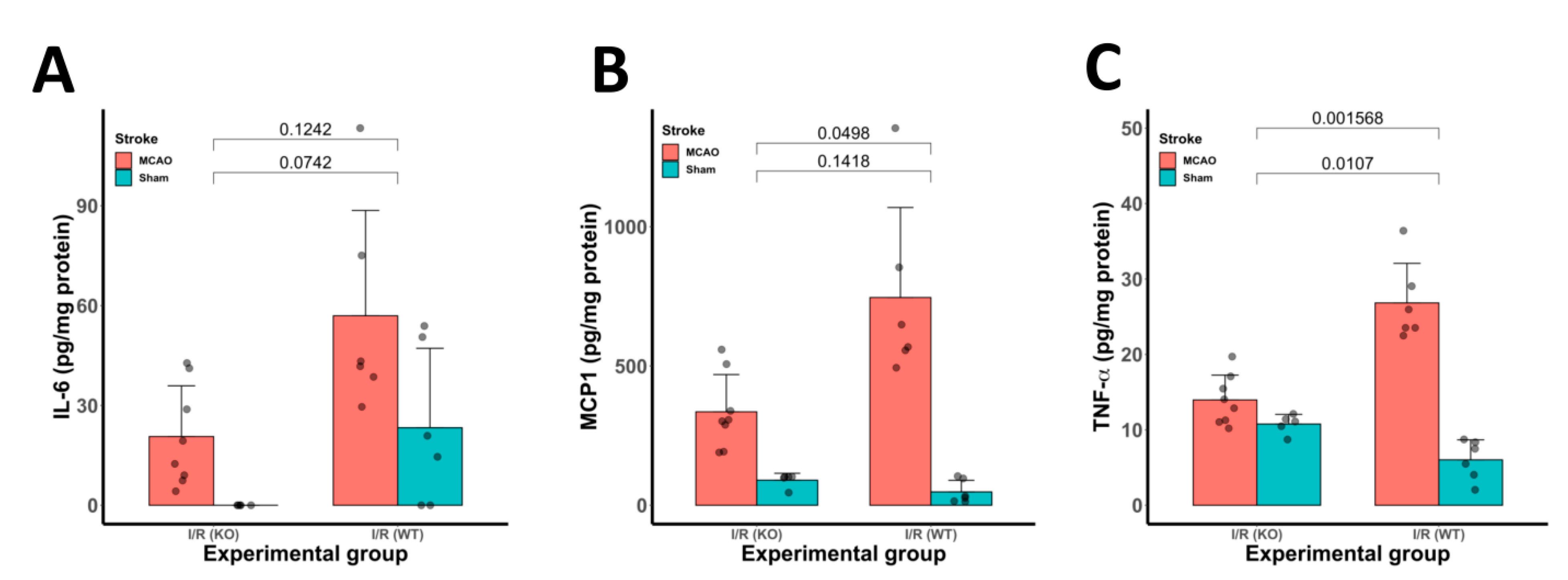
3.5. Inflammatory Cytokine Protein Expression Was Reduced in CYP2E1(−/−) Mice Following tMCAo
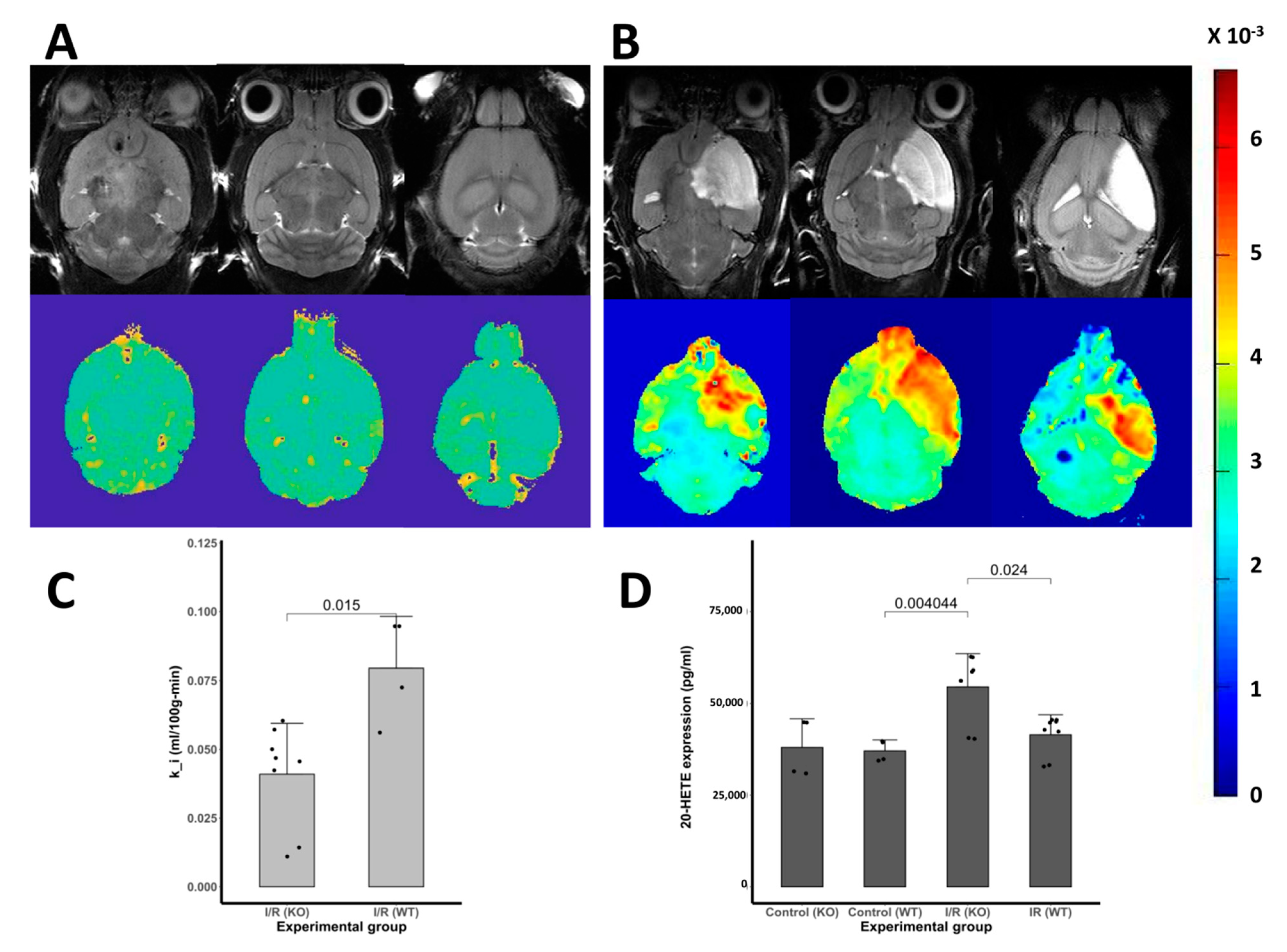
3.6. Blood-Brain Barrier Leakage
3.7. The 20–HETE Expression in the Brain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.; Yu, A.M.; Ward, J.M.; Krausz, K.W.; Akiyama, T.E.; Feigenbaum, L.; Gonzalez, F.J. The CYP2E1-humanized transgenic mouse: Role of CYP2E1 in acetaminophen hepatotoxicity. Drug Metab. Dispos. 2005, 33, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. 2005, 569, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 potentiation of LPS and TNFalpha-induced hepatotoxicity by mechanisms involving enhanced oxidative and nitrosative stress, activation of MAP kinases, and mitochondrial dysfunction. Genes Nutr. 2010, 5, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porubsky, P.R.; Meneely, K.M.; Scott, E.E. Structures of human cytochrome P-450 2E1. Insights into the binding of inhibitors and both small molecular weight and fatty acid substrates. J. Biol. Chem. 2008, 283, 33698–33707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, T.; Tindberg, N.; Ingelman-Sundberg, M.; Kohler, C. Regional distribution of ethanol-inducible cytochrome P450 IIE1 in the rat central nervous system. Neuroscience 1990, 34, 451–463. [Google Scholar] [CrossRef]
- Yadav, S.; Dhawan, A.; Singh, R.L.; Seth, P.K.; Parmar, D. Expression of constitutive and inducible cytochrome P450 2E1 in rat brain. Mol. Cell. Biochem. 2006, 286, 171–180. [Google Scholar] [CrossRef]
- Howard, L.A.; Miksys, S.; Hoffmann, E.; Mash, D.; Tyndale, R.F. Brain CYP2E1 is induced by nicotine and ethanol in rat and is higher in smokers and alcoholics. Br. J. Pharmacol. 2003, 138, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Farin, F.M.; Omiecinski, C.J. Regiospecific expression of cytochrome P-450s and microsomal epoxide hydrolase in human brain tissue. J. Toxicol. Environ. Health 1993, 40, 317–335. [Google Scholar] [CrossRef]
- Valencia-Olvera, A.C.; Moran, J.; Camacho-Carranza, R.; Prospero-Garcia, O.; Espinosa-Aguirre, J.J. CYP2E1 induction leads to oxidative stress and cytotoxicity in glutathione-depleted cerebellar granule neurons. Toxicol. In Vitro 2014, 28, 1206–1214. [Google Scholar] [CrossRef]
- Kelicen, P.; Tindberg, N. Lipopolysaccharide induces CYP2E1 in astrocytes through MAP kinase kinase-3 and C/EBPbeta and -delta. J. Biol. Chem. 2004, 279, 15734–15742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tindberg, N.; Baldwin, H.A.; Cross, A.J.; IngelmanSundberg, M. Induction of cytochrome P450 2E1 expression in rat and gerbil astrocytes by inflammatory factors and ischemic injury. Mol. Pharmacol. 1996, 50, 1065–1072. [Google Scholar] [PubMed]
- Garcia-Suastegui, W.A.; Ramos-Chavez, L.A.; Rubio-Osornio, M.; Calvillo-Velasco, M.; Atzin-Mendez, J.A.; Guevara, J.; Silva-Adaya, D. The Role of CYP2E1 in the Drug Metabolism or Bioactivation in the Brain. Oxid. Med. Cell. Longev. 2017, 2017, 4680732. [Google Scholar] [CrossRef]
- Ohe, T.; Hirobe, M.; Mashino, T. Novel metabolic pathway of estrone and 17beta-estradiol catalyzed by cytochrome P-450. Drug Metab. Dispos. 2000, 28, 110–112. [Google Scholar] [PubMed]
- Miksys, S.; Tyndale, R.F. Cytochrome P450-mediated drug metabolism in the brain. J. Psychiatry Neurosci. JPN 2013, 38, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.; Galuppo, P.; Fraccarollo, D.; Vogt, C.; Widder, J.D.; Pfrang, J.; Tas, P.; Barbosa-Sicard, E.; Ruetten, H.; Ertl, G.; et al. Increased cytochrome P4502E1 expression and altered hydroxyeicosatetraenoic acid formation mediate diabetic vascular dysfunction: Rescue by guanylyl-cyclase activation. Diabetes 2010, 59, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Miyata, N.; Seki, T.; Tanaka, Y.; Omura, T.; Taniguchi, K.; Doi, M.; Bandou, K.; Kametani, S.; Sato, M.; Okuyama, S.; et al. Beneficial effects of a new 20-hydroxyeicosatetraenoic acid synthesis inhibitor, TS-011 [N-(3-chloro-4-morpholin-4-yl) phenyl-N’-hydroxyimido formamide], on hemorrhagic and ischemic stroke. J. Pharmacol. Exp. Ther. 2005, 314, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.L.; Du, J.; Zhang, Y.; Yan, J.T.; Hu, X.M. Hyperlipidemia exacerbates cerebral injury through oxidative stress, inflammation and neuronal apoptosis in MCAO/reperfusion rats. Exp. Brain Res. 2015, 233, 2753–2765. [Google Scholar] [CrossRef]
- Patel, S.A.; Bhambra, U.; Charalambous, M.P.; David, R.M.; Edwards, R.J.; Lightfoot, T.; Boobis, A.R.; Gooderham, N.J. Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br. J. Cancer 2014, 111, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, C.R.; Petrides, L.; Wilson, J.; Flatt, P.R.; Ioannides, C. Induction of rat hepatic mixed-function oxidases by acetone and other physiological ketones: Their role in diabetes-induced changes in cytochrome P450 proteins. Xenobiotica Fate Foreign Compd. Biol. Syst. 1992, 22, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhen, G.; Meloni, B.P.; Campbell, K.; Winn, H.R. Rodent Stroke Model Guidelines for Preclinical Stroke Trials (1st Edition). J. Exp. Stroke Transl. Med. 2009, 2, 2–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible Middle Cerebral-Artery Occlusion without Craniectomy in Rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miksys, S.; Hoffmann, E.; Tyndale, R.F. Regional and cellular induction of nicotine-metabolizing CYP2B1 in rat brain by chronic nicotine treatment. Biochem. Pharmacol. 2000, 59, 1501–1511. [Google Scholar] [CrossRef]
- Joshi, M.; Tyndale, R.F. Regional and cellular distribution of CYP2E1 in monkey brain and its induction by chronic nicotine. Neuropharmacology 2006, 50, 568–575. [Google Scholar] [CrossRef]
- Hartman, J.H.; Miller, G.P.; Meyer, J.N. Toxicological Implications of Mitochondrial Localization of CYP2E1. Toxicol. Res. 2017, 6, 273–289. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Methodology to assay CYP2E1 mixed function oxidase catalytic activity and its induction. Redox Biol. 2014, 2, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Koop, D.R.; Laethem, C.L.; Tierney, D.J. The utility of p-nitrophenol hydroxylation in P450IIE1 analysis. Drug Metab. Rev. 1989, 20, 541–551. [Google Scholar] [CrossRef]
- Ledesma, J.C.; Miquel, M.; Pascual, M.; Guerri, C.; Aragon, C.M. Induction of brain cytochrome P450 2E1 boosts the locomotor-stimulating effects of ethanol in mice. Neuropharmacology 2014, 85, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Islas, A.; Chagoya-Hazas, V.; Perez-Aguilar, B.; Palestino-Dominguez, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Gomez-Quiroz, L.E.; Gutierrez-Ruiz, M.C. Cholesterol Enhances the Toxic Effect of Ethanol and Acetaldehyde in Primary Mouse Hepatocytes. Oxidative Med. Cell. Longev. 2016, 2016, 9209825. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, H.; Gattoni-Celli, S.; Taheri, S.; Kindy, M.S. Dietary supplementation of GrandFusion((R)) mitigates cerebral ischemia-induced neuronal damage and attenuates inflammation. Nutr. Neurosci. 2016, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Shinomol, G.K.; Muralidhara. Effect of Centella asiatica leaf powder on oxidative markers in brain regions of prepubertal mice in vivo and its in vitro efficacy to ameliorate 3-NPA-induced oxidative stress in mitochondria. Phytomedicine 2008, 15, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Baltes, C.; Radzwill, N.; Bosshard, S.; Marek, D.; Rudin, M. Micro MRI of the mouse brain using a novel 400 MHz cryogenic quadrature RF probe. NMR Biomed. 2009, 22, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Candelario-Jalil, E.; Estrada, E.Y.; Rosenberg, G.A. Spatiotemporal correlations between blood-brain barrier permeability and apparent diffusion coefficient in a rat model of ischemic stroke. PLoS ONE 2009, 4, e6597. [Google Scholar] [CrossRef]
- Pan, J.; Konstas, A.A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Y.; Liu, J.; Li, Y.; Li, B.; Zhang, Y.Y.; Jing, Z.W.; Yu, Y.N.; Li, H.X.; Guo, S.S.; Zhao, Y.J.; et al. Time-dependent variation of pathways and networks in a 24-hour window after cerebral ischemia-reperfusion injury. BMC Syst. Biol. 2015, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Knockaert, L.; Descatoire, V.; Vadrot, N.; Fromenty, B.; Robin, M.A. Mitochondrial CYP2E1 is sufficient to mediate oxidative stress and cytotoxicity induced by ethanol and acetaminophen. Toxicol. In Vitro 2011, 25, 475–484. [Google Scholar] [CrossRef]
- Walther, B.; Ghersi-Egea, J.F.; Minn, A.; Siest, G. Subcellular distribution of cytochrome P-450 in the brain. Brain Res. 1986, 375, 338–344. [Google Scholar] [CrossRef]
- Iscan, M.; Reuhl, K.; Weiss, B.; Maines, M.D. Regional and subcellular distribution of cytochrome P-450-dependent drug metabolism in monkey brain: The olfactory bulb and the mitochondrial fraction have high levels of activity. Biochem. Biophys. Res. Commun. 1990, 169, 858–863. [Google Scholar] [CrossRef]
- Neve, E.P.; Ingelman-Sundberg, M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J. Biol. Chem. 2000, 275, 17130–17135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussadia, B.; Ghosh, C.; Plaud, C.; Pascussi, J.M.; de Bock, F.; Rousset, M.C.; Janigro, D.; Marchi, N. Effect of status epilepticus and antiepileptic drugs on CYP2E1 brain expression. Neuroscience 2014, 281, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulla, D.; Goralski, K.B.; Renton, K.W. The regulation of cytochrome P450 2E1 during LPS-induced inflammation in the rat. Toxicol. Appl. Pharmacol. 2006, 216, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Dong, G.; Luo, H.; Cao, J.; Wang, C.; Wu, J.; Feng, Y.Q.; Yue, J. Induction of brain CYP2E1 by chronic ethanol treatment and related oxidative stress in hippocampus, cerebellum, and brainstem. Toxicology 2012, 302, 275–284. [Google Scholar] [CrossRef]
- Roberts, B.J.; Shoaf, S.E.; Jeong, K.S.; Song, B.J. Induction of CYP2E1 in liver, kidney, brain and intestine during chronic ethanol administration and withdrawal: Evidence that CYP2E1 possesses a rapid phase half-life of 6 hours or less. Biochem. Biophys. Res. Commun. 1994, 205, 1064–1071. [Google Scholar] [CrossRef]
- Joshi, M.; Tyndale, R.F. Induction and recovery time course of rat brain CYP2E1 after nicotine treatment. Drug Metab. Dispos. 2006, 34, 647–652. [Google Scholar] [CrossRef] [Green Version]
- Wei, E.P.; Lamb, R.G.; Kontos, H.A. Increased phospholipase C activity after experimental brain injury. J. Neurosurg. 1982, 56, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.S.; Tyndale, R.F. Cytochrome P450 enzymes in the brain: Emerging evidence of biological significance. Trends Pharmacol. Sci. 2011, 32, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Kontos, C.D.; Wei, E.P.; Williams, J.I.; Kontos, H.A.; Povlishock, J.T. Cytochemical detection of superoxide in cerebral inflammation and ischemia in vivo. Am. J. Physiol. 1992, 263, H1234–H1242. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Juurlink, B.H. Response of glial cells to ischemia: Roles of reactive oxygen species and glutathione. Neurosci. Biobehav. Rev. 1997, 21, 151–166. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Schroeter, M. Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 1998, 56, 149–171. [Google Scholar] [CrossRef]
- Jin, G.; Arai, K.; Murata, Y.; Wang, S.; Stins, M.F.; Lo, E.H.; van Leyen, K. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 2008, 39, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario-Jalil, E.; Taheri, S.; Yang, Y.; Sood, R.; Grossetete, M.; Estrada, E.Y.; Fiebich, B.L.; Rosenberg, G.A. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J. Pharmacol. Exp. Ther. 2007, 323, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox. Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Rust, R.; Gronnert, L.; Schwab, M.E. Inflammation after Stroke: A Local Rather Than Systemic Response? Trends Neurosci. 2018, 41, 877–879. [Google Scholar] [CrossRef]
- Ng, G.Y.; Lim, Y.A.; Sobey, C.G.; Dheen, T.; Fann, D.Y.; Arumugam, T.V. Epigenetic regulation of inflammation in stroke. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418771815. [Google Scholar] [CrossRef]
- Martin, A.; Domercq, M.; Matute, C. Inflammation in stroke: The role of cholinergic, purinergic and glutamatergic signaling. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774267. [Google Scholar] [CrossRef] [Green Version]
- Wood, H. Stroke: An alarmin’ inflammatory response after stroke. Nat. Rev. Neurol. 2018, 14, 252–253. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, G.W.; van Ess, P.J.; Watson, A.M.; Mattson, M.P.; Blouin, R.A. Cytochrome P450 and antioxidant activity in interleukin-6 knockout mice after induction of the acute-phase response. J. Interferon Cytokine Res. 2001, 21, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Elshenawy, O.H.; Shoieb, S.M.; Mohamed, A.; El-Kadi, A.O. Clinical Implications of 20-Hydroxyeicosatetraenoic Acid in the Kidney, Liver, Lung and Brain: An Emerging Therapeutic Target. Pharmaceutics 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 2018, 833, 190–200. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Omura, T.; Tanaka, Y.; Miyata, N.; Koizumi, C.; Sakurai, T.; Fukasawa, M.; Hachiuma, K.; Minagawa, T.; Susumu, T.; Yoshida, S.; et al. Effect of a new inhibitor of the synthesis of 20-HETE on cerebral ischemia reperfusion injury. Stroke 2006, 37, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Renic, M.; Klaus, J.A.; Omura, T.; Kawashima, N.; Onishi, M.; Miyata, N.; Koehler, R.C.; Harder, D.R.; Roman, R.J. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 2009, 29, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals Through G-Protein-Coupled Receptor GPR75 (G(q)) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Zhu, H.; Kindy, M.S.; Taheri, S. Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury. Antioxidants 2021, 10, 52. https://doi.org/10.3390/antiox10010052
Yu J, Zhu H, Kindy MS, Taheri S. Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury. Antioxidants. 2021; 10(1):52. https://doi.org/10.3390/antiox10010052
Chicago/Turabian StyleYu, Jin, Hong Zhu, Mark S. Kindy, and Saeid Taheri. 2021. "Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury" Antioxidants 10, no. 1: 52. https://doi.org/10.3390/antiox10010052
APA StyleYu, J., Zhu, H., Kindy, M. S., & Taheri, S. (2021). Cytochrome P450 CYP2E1 Suppression Ameliorates Cerebral Ischemia Reperfusion Injury. Antioxidants, 10(1), 52. https://doi.org/10.3390/antiox10010052