
Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose


 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Apoptosis Assay
2.3. Cell Viability Assay
2.4. RNA Isolation and Quantitative Real-Time PCR
2.5. Measurement of Intracellular ROS
2.6. Cloning of the p47-roGFP Biosensor Construct and Production of Lentiviral Particles
2.7. Generation of HREC/p47roGFP Stable Cell Line
2.8. Determination of NOX2-Associated ROS Using NOX-Specific Redox Biosensor p47-roGFP
2.9. Protein Quantification by ELISA
2.10. Statistical Analysis
3. Results and Discussion
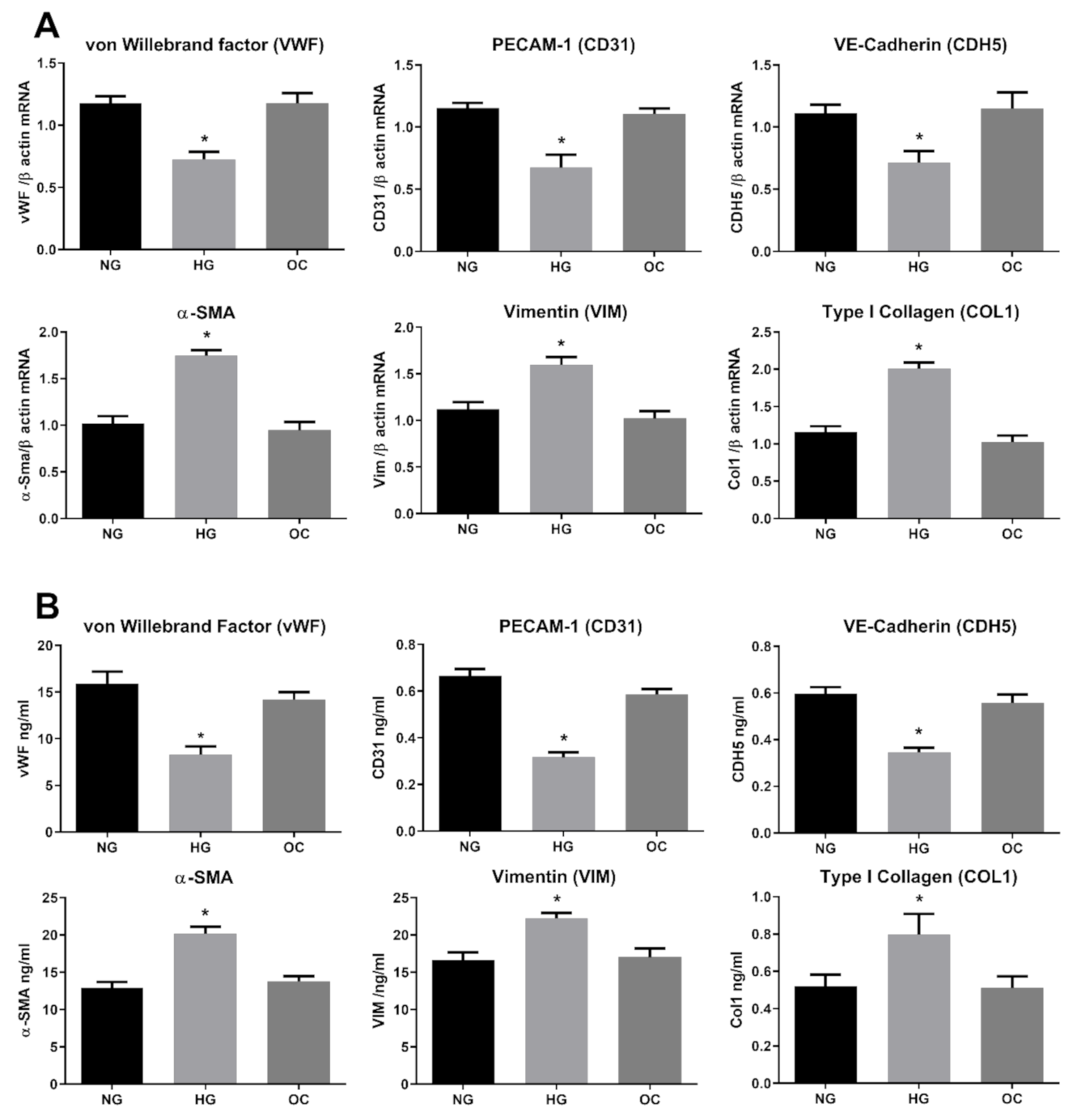
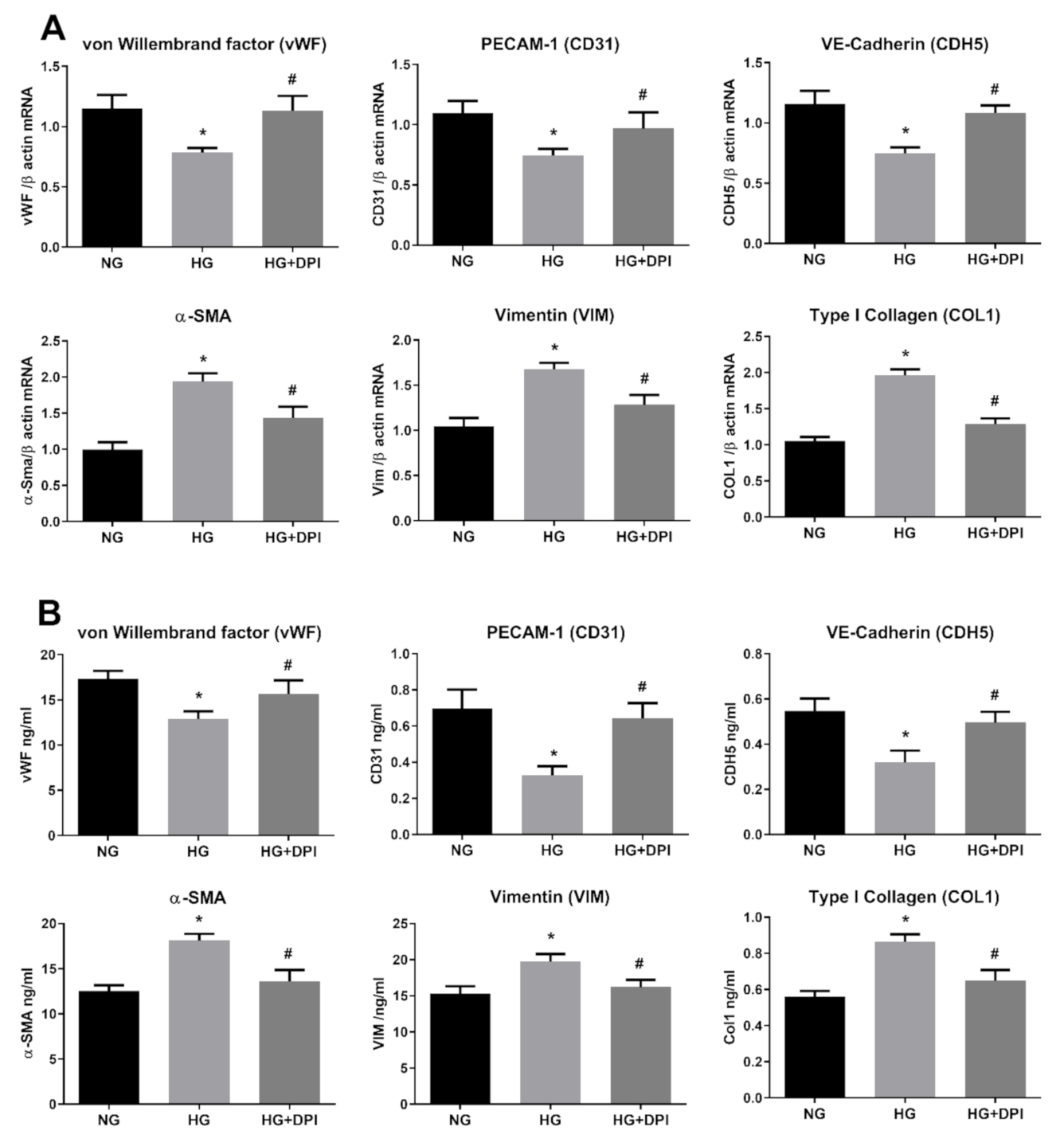
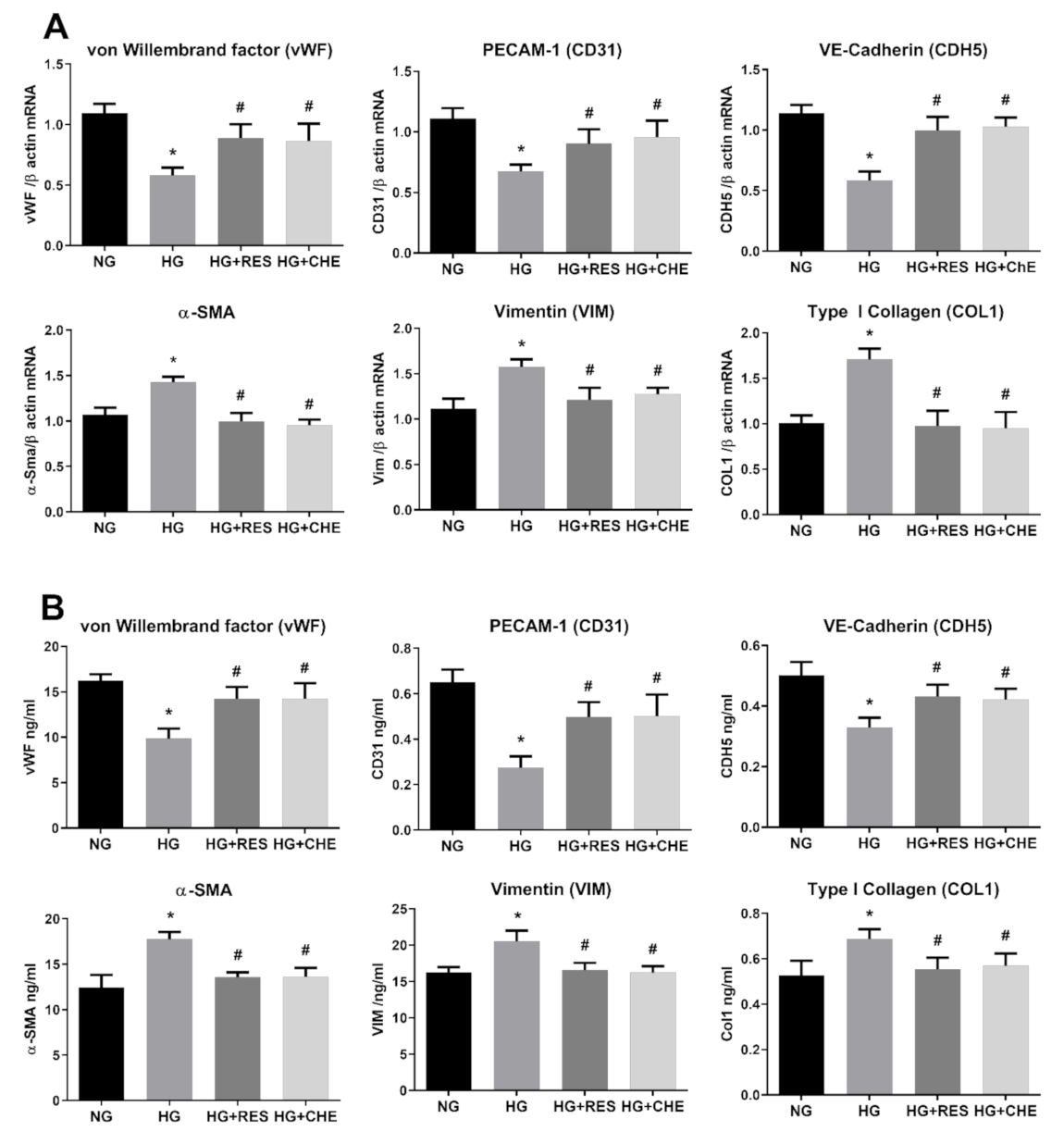
3.1. High Glucose Induces EndMT in HRECs
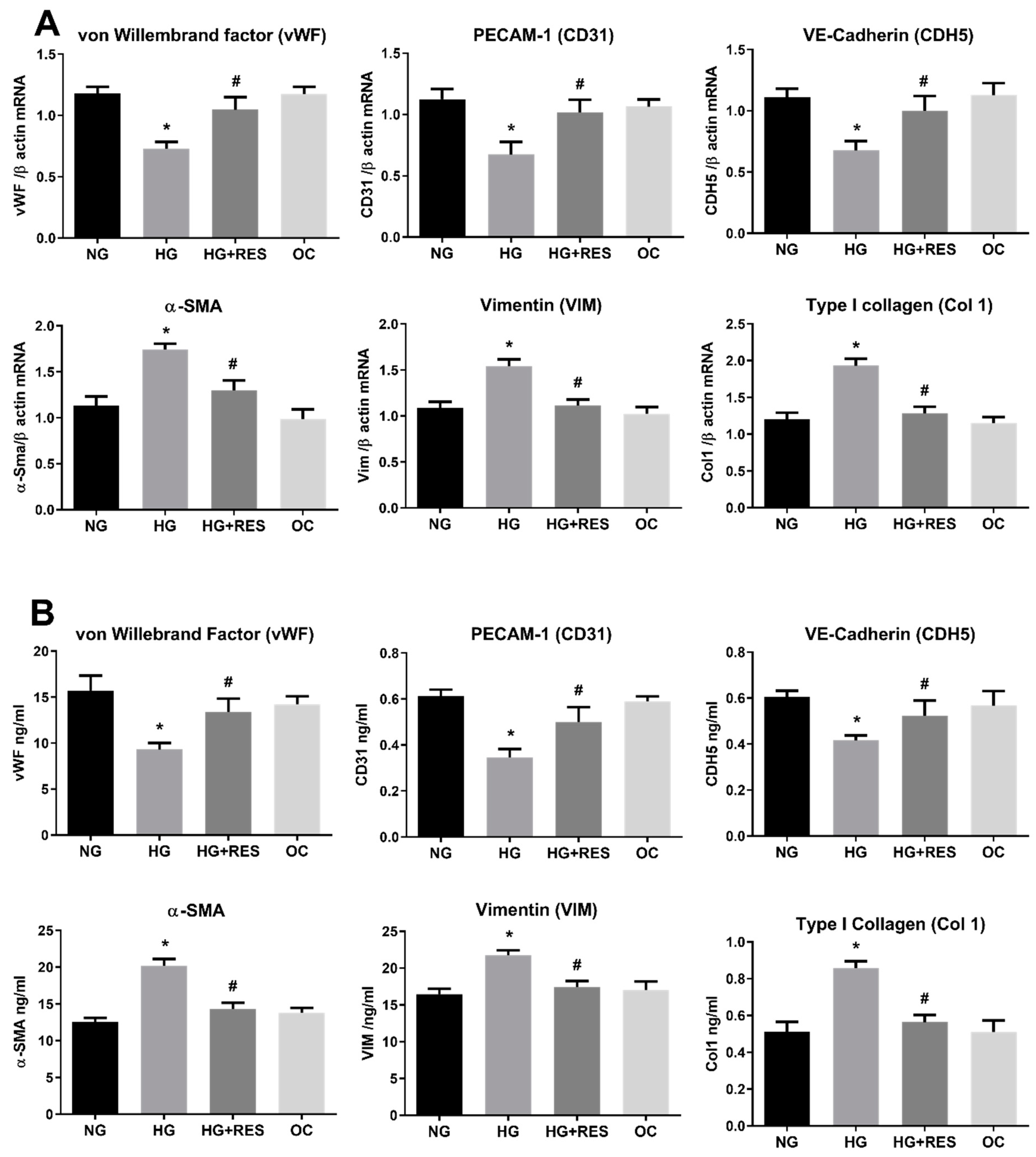
3.2. Resveratrol Counteracts HG-Induced EndMT
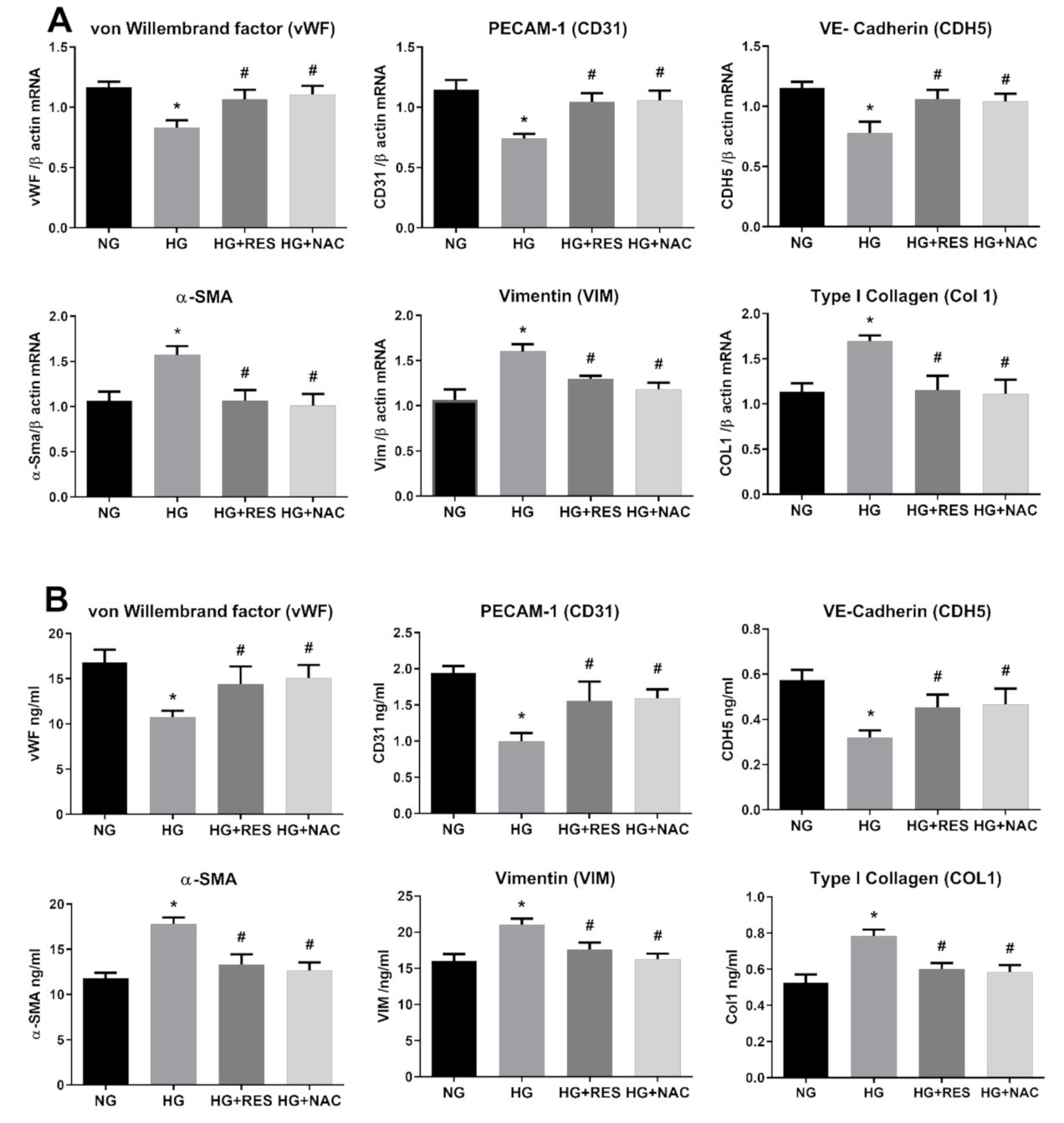
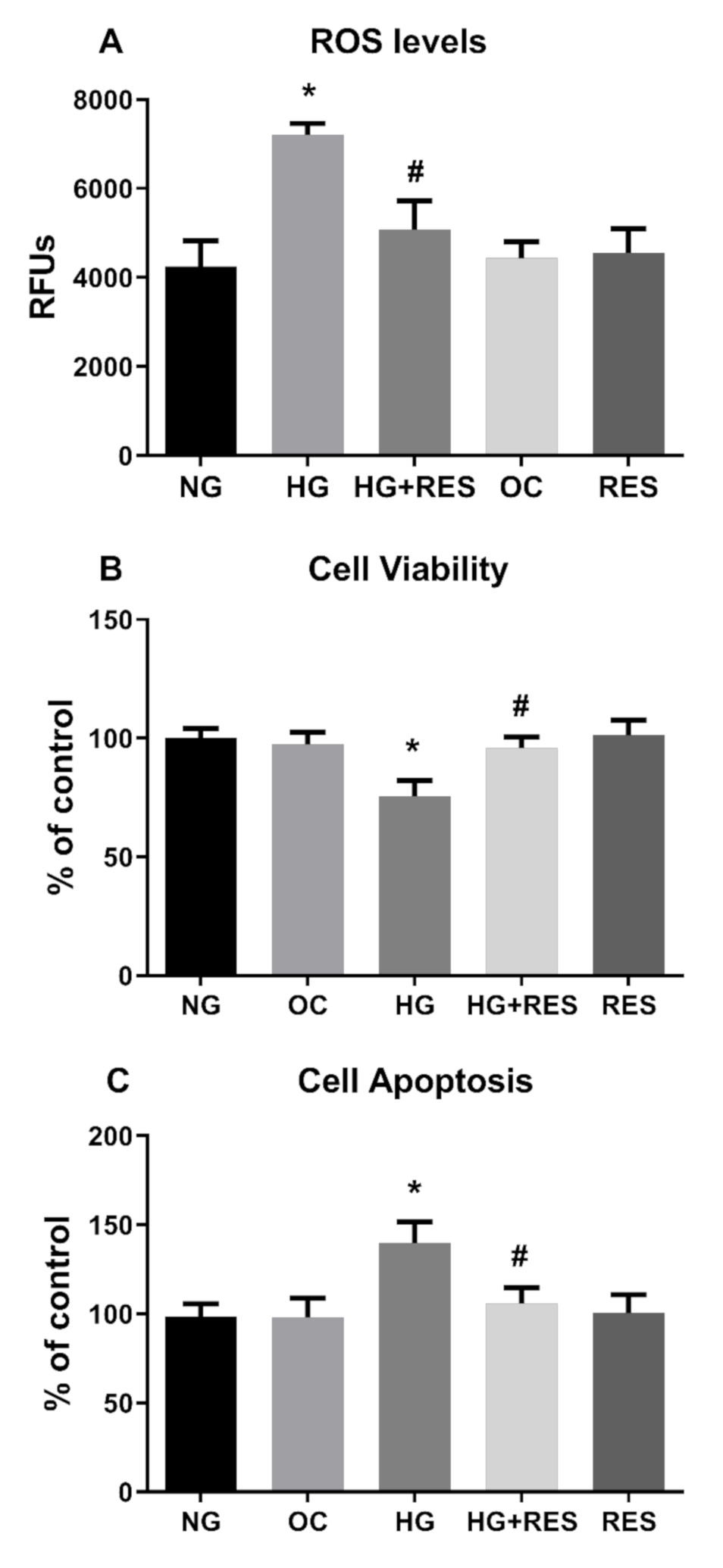
3.3. Resveratrol Counteracts HG-Induced ROS Generation and EndMT in HRECs
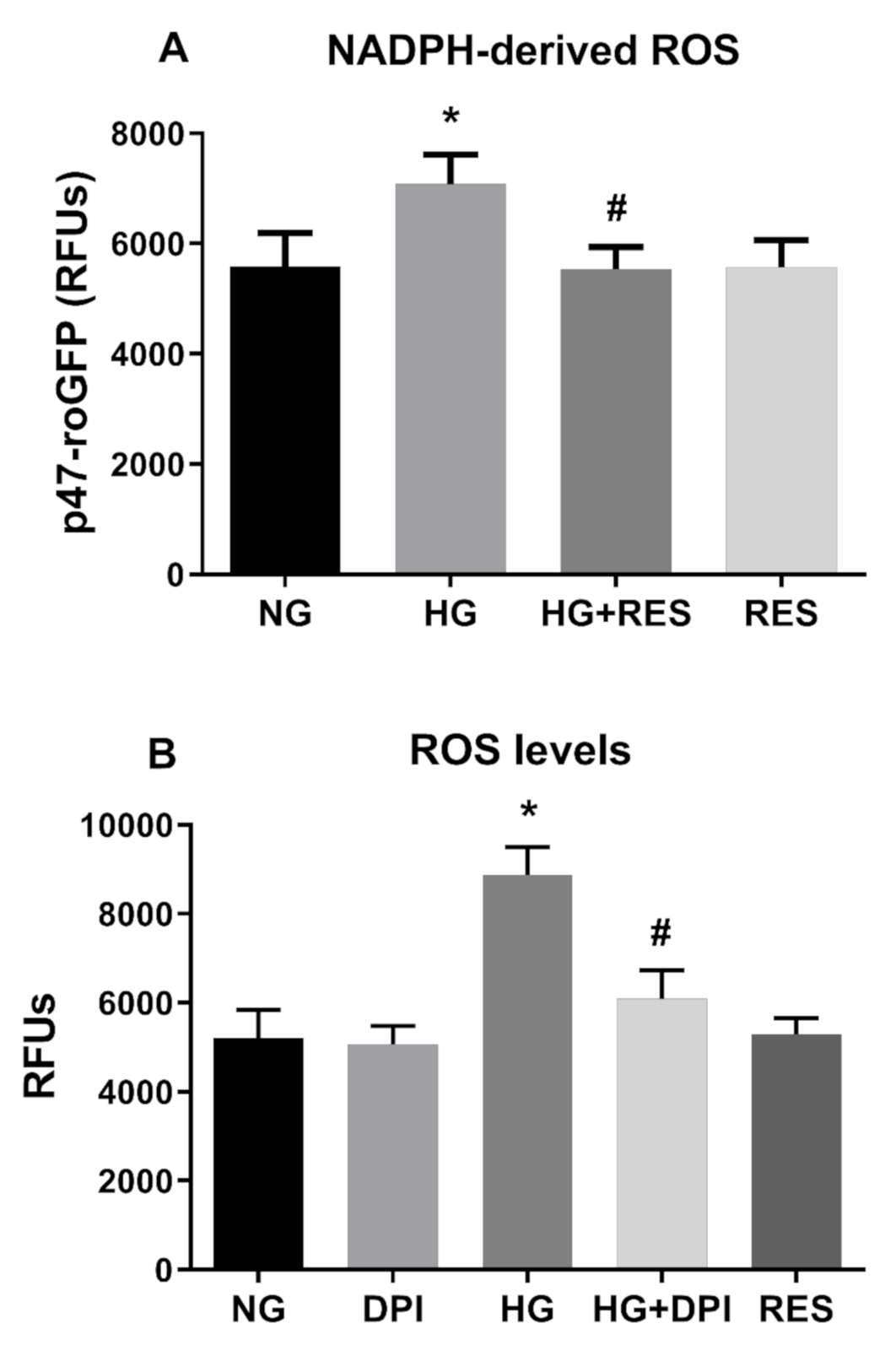
3.4. NADPH Oxidases Mediates HG-Induced ROS Generation
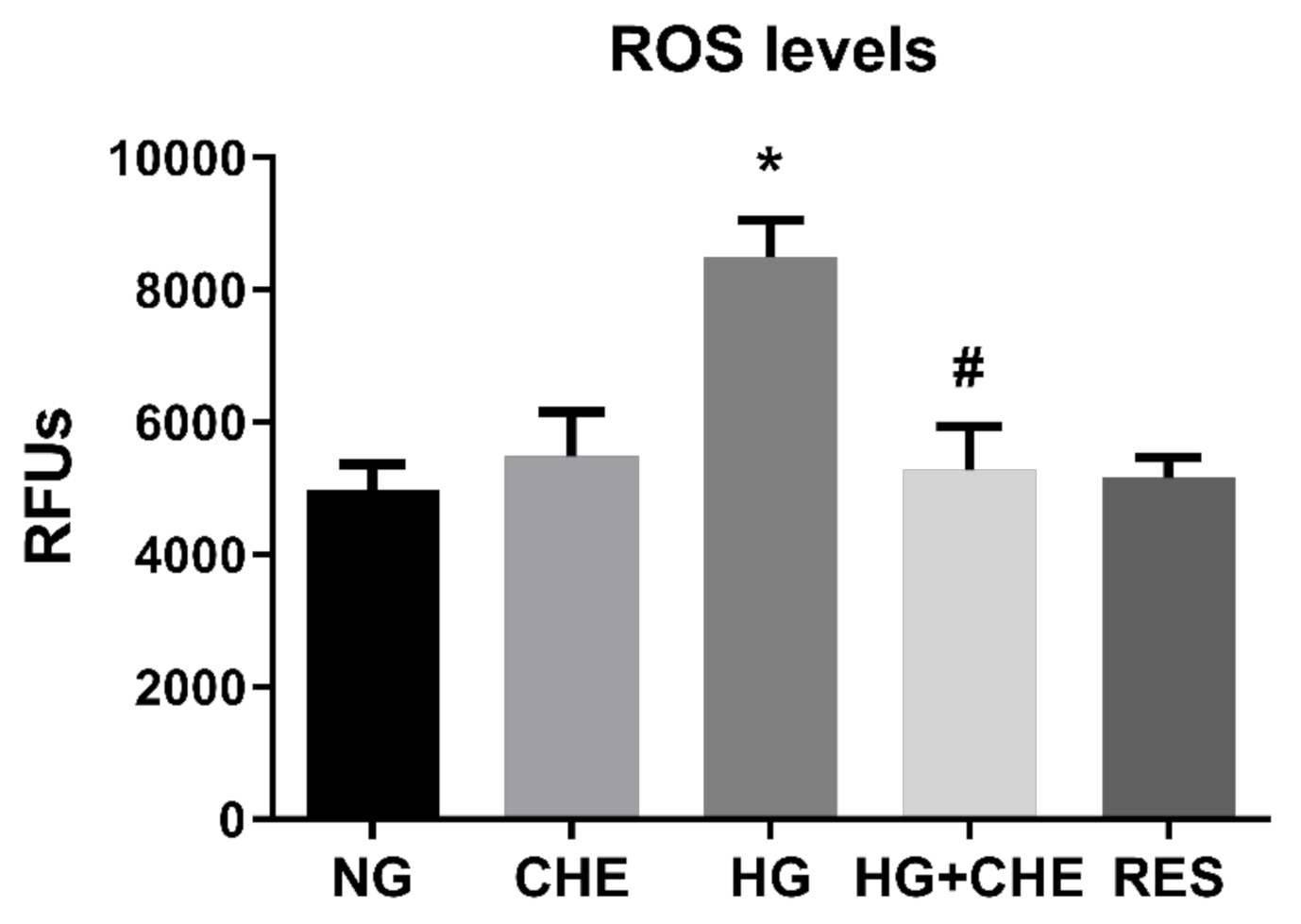
3.5. HG Activates NADPH Via PKC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kharroubi, A.T.; Darwish, H.M. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Gordin, D.; Groop, P.H. Aspects of Hyperglycemia Contribution to Arterial Stiffness and Cardiovascular Complications in Patients with Type 1 Diabetes. J. Diabetes Sci. Technol. 2016, 10, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.R.; Twigg, S.M. Fibrosis in diabetes complications: Pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008, 4, 575–596. [Google Scholar]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bernatchez, P.N.; de Haan, J.B. Targeting endothelial dysfunction in vascular complications associated with diabetes. Int. J. Vasc. Med. 2012, 2012, 750126. [Google Scholar] [CrossRef]
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy-ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Hong, L.; Du, X.; Li, W.; Mao, Y.; Sun, L.; Li, X. EndMT: A promising and controversial field. Eur. J. Cell Biol. 2018, 97, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.P. Hyperglycemic endothelial dysfunction: Does it happen and does it matter? J. Thorac. Dis. 2015, 7, 1693. [Google Scholar] [PubMed]
- Peng, H.; Li, Y.; Wang, C.; Zhang, J.; Chen, Y.; Chen, W.; Cao, J.; Wang, Y.; Hu, Z.; Lou, T. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 2016, 6, 20304. [Google Scholar] [CrossRef]
- Liu, X.; Mujahid, H.; Rong, B.; Lu, Q.H.; Zhang, W.; Li, P.; Li, N.; Liang, E.S.; Wang, Q.; Tang, D.Q.; et al. Irisin inhibits high glucose-induced endothelial-to-mesenchymal transition and exerts a dose-dependent bidirectional effect on diabetic cardiomyopathy. J. Cell. Mol. Med. 2018, 22, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Feng, B.; Chen, S.; Chu, Y.; Chakrabarti, S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7321–7331. [Google Scholar] [CrossRef] [Green Version]
- El-Asrar, A.M.A. Endothelial-to-mesenchymal transition contributes to the myofibroblast population in proliferative diabetic retinopathy. Saudi J. Ophthalmol. 2016, 30, 1. [Google Scholar] [CrossRef] [Green Version]
- El-Asrar, A.M.A.; De Hertogh, G.; van den Eynde, K.; Alam, K.; Van Raemdonck, K.; Opdenakker, G.; Van Damme, J.; Geboes, K.; Struyf, S. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Exp. Eye Res. 2015, 132, 179–189. [Google Scholar] [CrossRef]
- Chang, W.; Lajko, M.; Fawzi, A.A. Endothelin-1 is associated with fibrosis in proliferative diabetic retinopathy membranes. PLoS ONE 2018, 13, e0191285. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Inoguchi, T. The role of oxidative stress in the pathogenesis of diabetic vascular complications. Diabetes Metab. J. 2012, 36, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jimenez-Altayó, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2020, 20. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef] [PubMed]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [Green Version]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD (P) H oxidase mediates TGF-β1–induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Li, G.N.; Xie, J.; Li, R.; Chen, Q.H.; Chen, J.Z.; Wei, Z.H.; Kang, L.N.; Xu, B. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-beta/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc. Disord. 2016, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Chen, Y.H.; Tai, M.C.; Liang, C.M.; Lu, D.W.; Chen, J.T. Resveratrol inhibits transforming growth factor-beta2-induced epithelial-to-mesenchymal transition in human retinal pigment epithelial cells by suppressing the Smad pathway. Drug Des. Dev. Ther. 2017, 11, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinellu, A.; Sotgia, S.; Scanu, B.; Pintus, G.; Posadino, A.M.; Cossu, A.; Deiana, L.; Sengupta, S.; Carru, C. S-homocysteinylated LDL apolipoprotein B adversely affects human endothelial cells in vitro. Atherosclerosis 2009, 206, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Floris, I.; Descamps, B.; Vardeu, A.; Mitić, T.; Posadino, A.M.; Shantikumar, S.; Sala-Newby, G.; Capobianco, G.; Mangialardi, G.; Howard, L. Gestational diabetes mellitus impairs fetal endothelial cell functions through a mechanism involving microRNA-101 and histone methyltransferase enhancer of zester homolog-2. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, C.; Pintus, G.; Fiori, M.G.; Bennardini, F.; Pinna, G.; Gaspa, L. Opioid Peptide Gene Expression in the Primary Hereditary Cardiomyopathy of the Syrian Hamster, I. Regulation of Prodynorphin Gene Expression by Nuclear Protein Kinase C. J. Biol. Chem. 1997, 272, 6685–6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, C.; Maioli, M.; Pintus, G.; Gottardi, G.; Bersani, F. Elf-pulsed magnetic fields modulate opioid peptide gene expression in myocardial cells. Cardiovasc. Res. 2000, 45, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Debidda, M.; Sanna, B.; Cossu, A.; Posadino, A.M.; Tadolini, B.; Ventura, C.; Pintus, G. NAMI-A inhibits the PMA-induced ODC gene expression in ECV304 cells: Involvement of PKC/Raf/Mek/ERK signalling pathway. Int. J. Oncol. 2003, 23, 477–482. [Google Scholar] [CrossRef]
- Pasciu, V.; Posadino, A.M.; Cossu, A.; Sanna, B.; Tadolini, B.; Gaspa, L.; Marchisio, A.; Dessole, S.; Capobianco, G.; Pintus, G. Akt downregulation by flavin oxidase–induced ROS generation mediates dose-dependent endothelial cell damage elicited by natural antioxidants. Toxicol. Sci. 2010, 114, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Nakagawa, K.; Tsuduki, T.; Miyazawa, T. α-Tocopherol suppresses antiangiogenic effect of δ-tocotrienol in human umbilical vein endothelial cells. J. Nutr. Biochem. 2015, 26, 345–350. [Google Scholar] [CrossRef]
- Posadino, A.M.; Giordo, R.; Cossu, A.; Nasrallah, G.K.; Shaito, A.; Abou-Saleh, H.; Eid, A.H.; Pintus, G. Flavin oxidase-induced ROS generation modulates PKC biphasic effect of resveratrol on endothelial cell survival. Biomolecules 2019, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Posadino, A.M.; Biosa, G.; Zayed, H.; Abou-Saleh, H.; Cossu, A.; Nasrallah, G.K.; Giordo, R.; Pagnozzi, D.; Porcu, M.C.; Pretti, L. Protective effect of cyclically pressurized solid–liquid extraction polyphenols from Cagnulari grape pomace on oxidative endothelial cell death. Molecules 2018, 23, 2105. [Google Scholar] [CrossRef] [Green Version]
- Vono, R.; Fuoco, C.; Testa, S.; Pirrò, S.; Maselli, D.; McCollough, D.F.; Sangalli, E.; Pintus, G.; Giordo, R.; Finzi, G. Activation of the pro-oxidant PKCβII-p66Shc signaling pathway contributes to pericyte dysfunction in skeletal muscles of patients with diabetes with critical limb ischemia. Diabetes 2016, 65, 3691–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintus, G.; Giordo, R.; Wang, Y.; Zhu, W.; Kim, S.H.; Zhang, L.; Ni, L.; Zhang, J.; Telljohann, R.; McGraw, K.R. Reduced vasorin enhances angiotensin II signaling within the aging arterial wall. Oncotarget 2018, 9, 27117. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Deiana, L.; Carru, C.; Pintus, G. Coumaric acid induces mitochondrial damage and oxidative-mediated cell death of human endothelial cells. Cardiovasc. Toxicol. 2013, 13, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Begemann, S.; Galimi, F.; Karlseder, J. Moderate expression of TRF2 in the hematopoietic system increases development of large cell blastic T-cell lymphomas. Aging 2009, 1, 122–130. [Google Scholar] [CrossRef]
- Dissen, G.A.; Lomniczi, A.; Neff, T.L.; Hobbs, T.R.; Kohama, S.G.; Kroenke, C.D.; Galimi, F.; Ojeda, S.R. In vivo manipulation of gene expression in non-human primates using lentiviral vectors as delivery vehicles. Methods 2009, 49, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Carru, C.; Pintus, G. Resveratrol alters human endothelial cells redox state and causes mitochondrial-dependent cell death. Food Chem. Toxicol. 2015, 78, 10–16. [Google Scholar] [CrossRef]
- Cossu, A.; Posadino, A.M.; Giordo, R.; Emanueli, C.; Sanguinetti, A.M.; Piscopo, A.; Poiana, M.; Capobianco, G.; Piga, A.; Pintus, G. Apricot melanoidins prevent oxidative endothelial cell death by counteracting mitochondrial oxidation and membrane depolarization. PLoS ONE 2012, 7, e48817. [Google Scholar] [CrossRef] [Green Version]
- Boin, F.; Erre, G.L.; Posadino, A.M.; Cossu, A.; Giordo, R.; Spinetti, G.; Passiu, G.; Emanueli, C.; Pintus, G. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet J. Rare Dis. 2014, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Fois, A.G.; Posadino, A.M.; Giordo, R.; Cossu, A.; Agouni, A.; Rizk, N.M.; Pirina, P.; Carru, C.; Zinellu, A.; Pintus, G. Antioxidant activity mediates pirfenidone antifibrotic effects in human pulmonary vascular smooth muscle cells exposed to sera of idiopathic pulmonary fibrosis patients. Oxid. Med. Cell. Longev. 2018, 2018, 2639081. [Google Scholar] [CrossRef]
- Yu, C.H.; Suriguga; Gong, M.; Liu, W.J.; Cui, N.X.; Wang, Y.; Du, X.; Yi, Z.C. High glucose induced endothelial to mesenchymal transition in human umbilical vein endothelial cell. Exp. Mol. Pathol. 2017, 102, 377–383. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair 2012, 5, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Resveratrol and ophthalmic diseases. Nutrients 2016, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Kim, Y.S.; Roh, G.S.; Choi, W.S.; Cho, G.J. Resveratrol blocks diabetes-induced early vascular lesions and vascular endothelial growth factor induction in mouse retinas. Acta Ophthalmol. 2012, 90, e31–e37. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xiong, J.; Nie, L.; Yu, Y.; Guan, X.; Xu, X.; Xiao, T.; Yang, K.; Liu, L.; Zhang, D.; et al. Resveratrol inhibits renal interstitial fibrosis in diabetic nephropathy by regulating AMPK/NOX4/ROS pathway. J. Mol. Med. 2016, 94, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Zhang, Y.; Wang, X.; Fan, X.-F.; Zhang, Y.; Li, X.; Gong, Y.-S.; Han, L.-P. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed. Pharmacother. 2019, 118, 109227. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Guan, X.; Wang, S.; Xiao, T.; Yang, K.; Xu, X.; Wang, J.; Zhao, J. Resveratrol prevents high glucose-induced epithelial-mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol. Cell. Endocrinol. 2015, 402, 13–20. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Zhang, T.; Zhou, Q.; Liu, J.; Liu, Y.; Kong, D.; Yu, W.; Liu, R.; Hai, C. TGF-beta1 induces epithelial-to-mesenchymal transition via inhibiting mitochondrial functions in A549 cells. Free Radic. Res. 2018, 52, 1432–1444. [Google Scholar] [CrossRef]
- Hao, Y.M.; Yuan, H.Q.; Ren, Z.; Qu, S.L.; Liu, L.S.; Dang, H.; Yin, K.; Fu, M.; Jiang, Z.S. Endothelial to mesenchymal transition in atherosclerotic vascular remodeling. Clin. Chim. Acta 2019, 490, 34–38. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Man, S.; Duffhues, G.S.; ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 22, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordo, R.; Cossu, A.; Pasciu, V.; Hoa, P.T.; Posadino, A.M.; Pintus, G. Different redox response elicited by naturally occurring antioxidants in human endothelial cells. Open Biochem. J. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purohit, V.; Simeone, D.M.; Lyssiotis, C.A. Metabolic regulation of redox balance in cancer. Cancers 2019, 11, 955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegrist, J.; Sies, H. Disturbed Redox Homeostasis in Oxidative Distress: A Molecular Link From Chronic Psychosocial Work Stress to Coronary Heart Disease? Circ. Res. 2017, 121, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelialmesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx2/Snail signaling pathways in MCF7 cells. Mol. Med. Rep. 2019, 20, 2339–2346. [Google Scholar] [CrossRef]
- Montorfano, I.; Becerra, A.; Cerro, R.; Echeverría, C.; Sáez, E.; Morales, M.G.; Fernández, R.; Cabello-Verrugio, C.; Simon, F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β 1 and TGF-β 2-dependent pathway. Lab. Investig. 2014, 94, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Posadino, A.M.; Phu, H.T.; Cossu, A.; Giordo, R.; Fois, M.; Thuan, D.T.B.; Piga, A.; Sotgia, S.; Zinellu, A.; Carru, C. Oxidative stress-induced Akt downregulation mediates green tea toxicity towards prostate cancer cells. Toxicol. In Vitro 2017, 42, 255–262. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [Green Version]
- Gorin, Y.; Block, K. Nox as a target for diabetic complications. Clin. Sci. 2013, 125, 361–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henríquez-Olguín, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to high-intensity interval training in skeletal muscle require NADPH oxidase 2. Redox Biol. 2019, 24, 101188. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Thakur, P.B.; Li, S.; Minard, C.; Rodney, G.G. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS ONE 2013, 8, e63989. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, T.; Dong, X. Advanced glycation end products of bovine serum albumin-induced endothelial-to-mesenchymal transition in cultured human and monkey endothelial cells via protein kinase B signaling cascades. Mol. Vision 2010, 16, 2669. [Google Scholar]
- Feng, L.; Zhang, C.; Liu, G.; Wang, F. RKIP negatively regulates the glucose induced angiogenesis and endothelial-mesenchymal transition in retinal endothelial cells. Exp. Eye Res. 2019, 189, 107851. [Google Scholar] [CrossRef]
- Gu, S.; Liu, Y.; Zou, J.; Wang, W.; Wei, T.; Wang, X.; Zhu, L.; Zhang, M.; Zhu, J.; Xie, T. Retinal pigment epithelial cells secrete miR-202-5p-containing exosomes to protect against proliferative diabetic retinopathy. Exp. Eye Res. 2020, 201, 108271. [Google Scholar] [CrossRef]
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. Suppl. 2007, 72, S49–S53. [Google Scholar] [CrossRef] [Green Version]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.A.; Chakrabarti, S. Cellular signaling and potential new treatment targets in diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 31867. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, e14–e22. [Google Scholar] [CrossRef] [PubMed]
- Gero, D. Hyperglycemia-induced endothelial dysfunction. In Endothelial Dysfunction–Old Concepts and New Challenges; Intechopen: London, UK, 2018; pp. 179–210. [Google Scholar]
- Engerman, R.; Bloodworth, J.; Nelson, S. Relationship of microvascular disease in diabetes to metabolic control. Diabetes 1977, 26, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Ghiselli, A.; Buchetti, B.; Carnevale, R.; Natella, F.; Germano, G.; Fimognari, F.; Di Santo, S.; Lenti, L.; Violi, F. Polyphenols synergistically inhibit oxidative stress in subjects given red and white wine. Atherosclerosis 2006, 188, 77–83. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordo, R.; Nasrallah, G.K.; Posadino, A.M.; Galimi, F.; Capobianco, G.; Eid, A.H.; Pintus, G. Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose. Antioxidants 2021, 10, 224. https://doi.org/10.3390/antiox10020224
Giordo R, Nasrallah GK, Posadino AM, Galimi F, Capobianco G, Eid AH, Pintus G. Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose. Antioxidants. 2021; 10(2):224. https://doi.org/10.3390/antiox10020224
Chicago/Turabian StyleGiordo, Roberta, Gheyath K. Nasrallah, Anna Maria Posadino, Francesco Galimi, Giampiero Capobianco, Ali Hussein Eid, and Gianfranco Pintus. 2021. "Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose" Antioxidants 10, no. 2: 224. https://doi.org/10.3390/antiox10020224
APA StyleGiordo, R., Nasrallah, G. K., Posadino, A. M., Galimi, F., Capobianco, G., Eid, A. H., & Pintus, G. (2021). Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose. Antioxidants, 10(2), 224. https://doi.org/10.3390/antiox10020224