
Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Brain Disorder and Mitochondrial Dysfunction
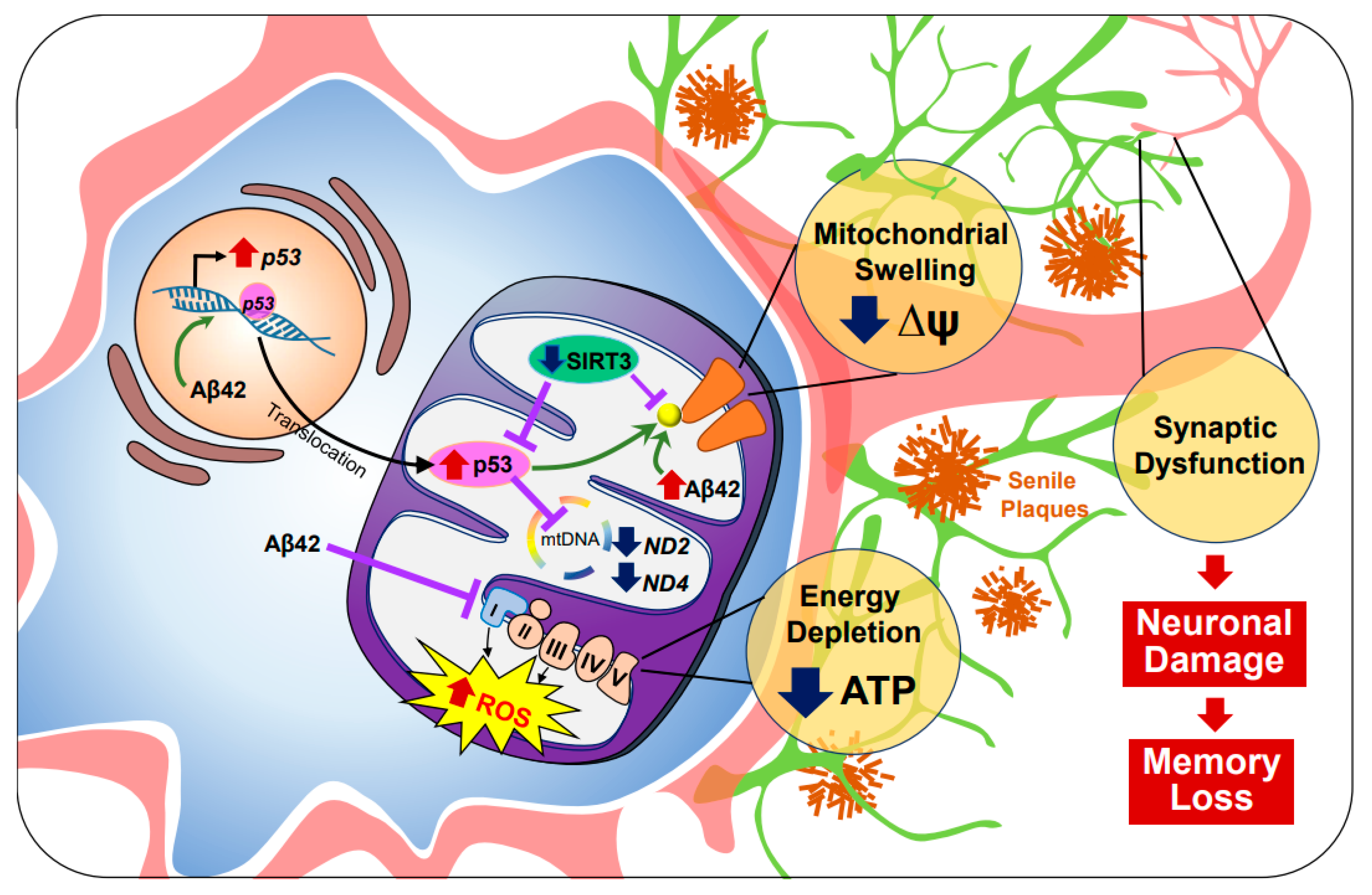
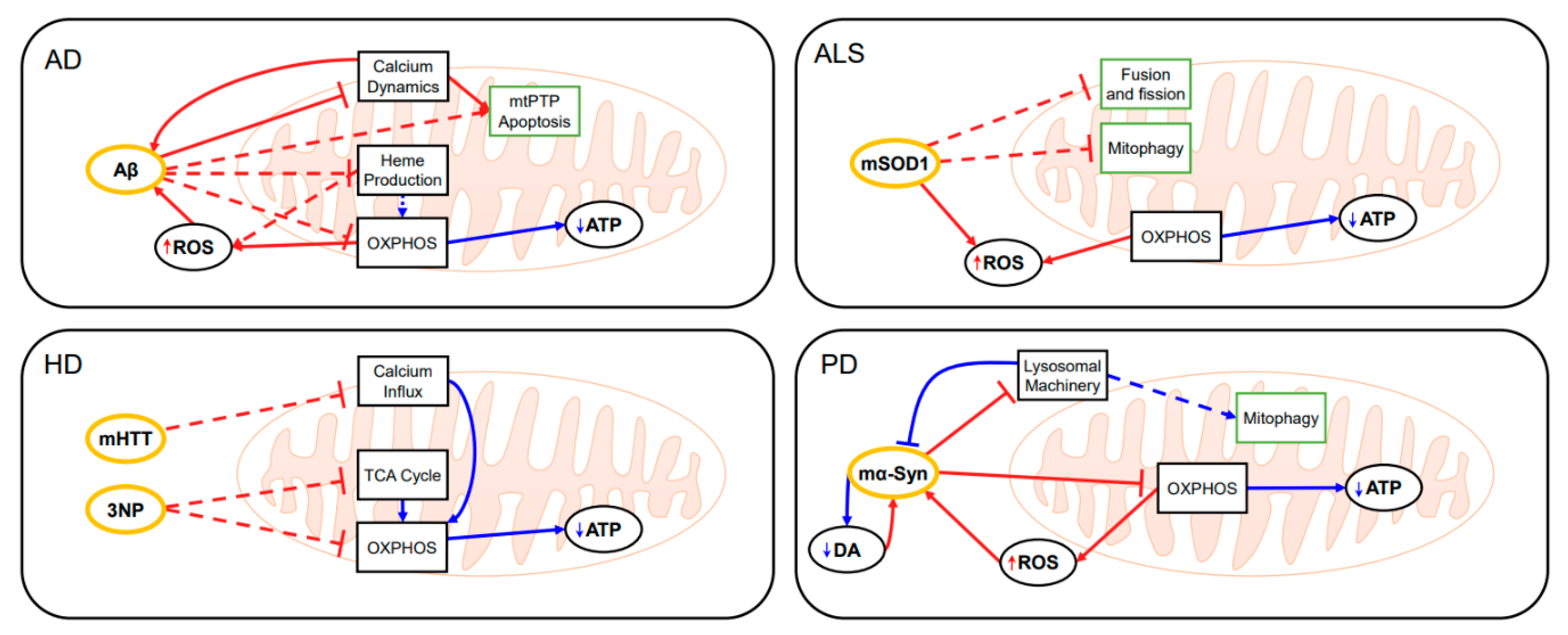
2.1. Alzheimer’s Disease (AD)
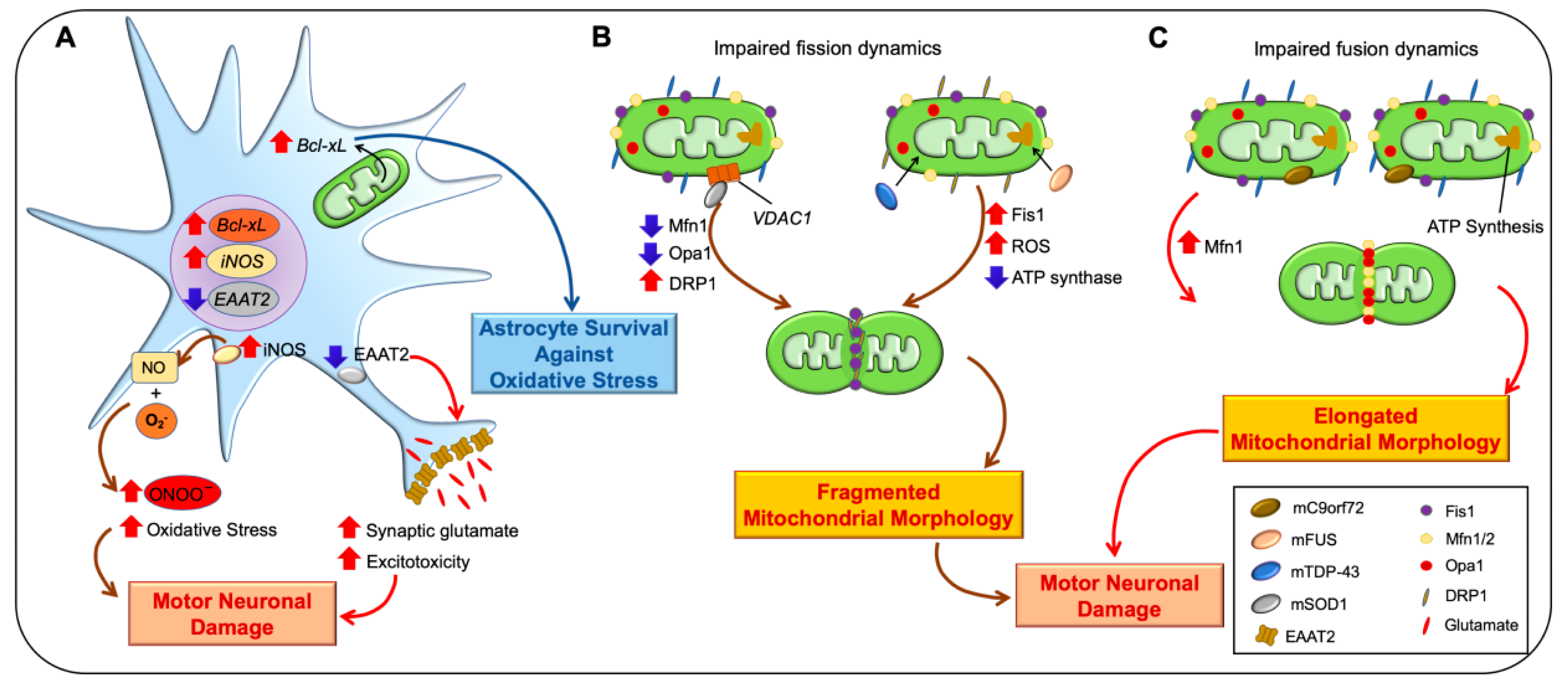
2.2. Amyotrophic Lateral Sclerosis (ALS)
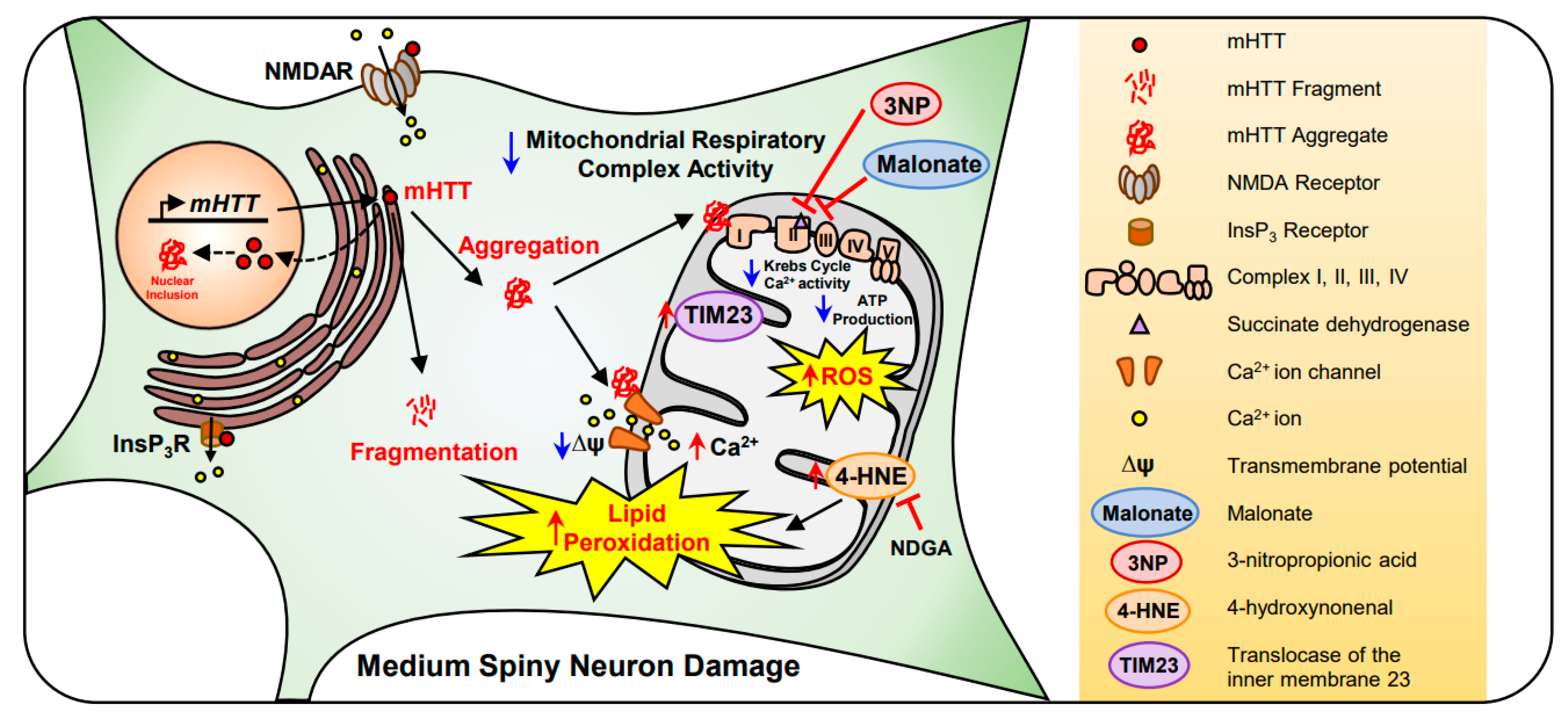
2.3. Huntington’s Disease (HD)
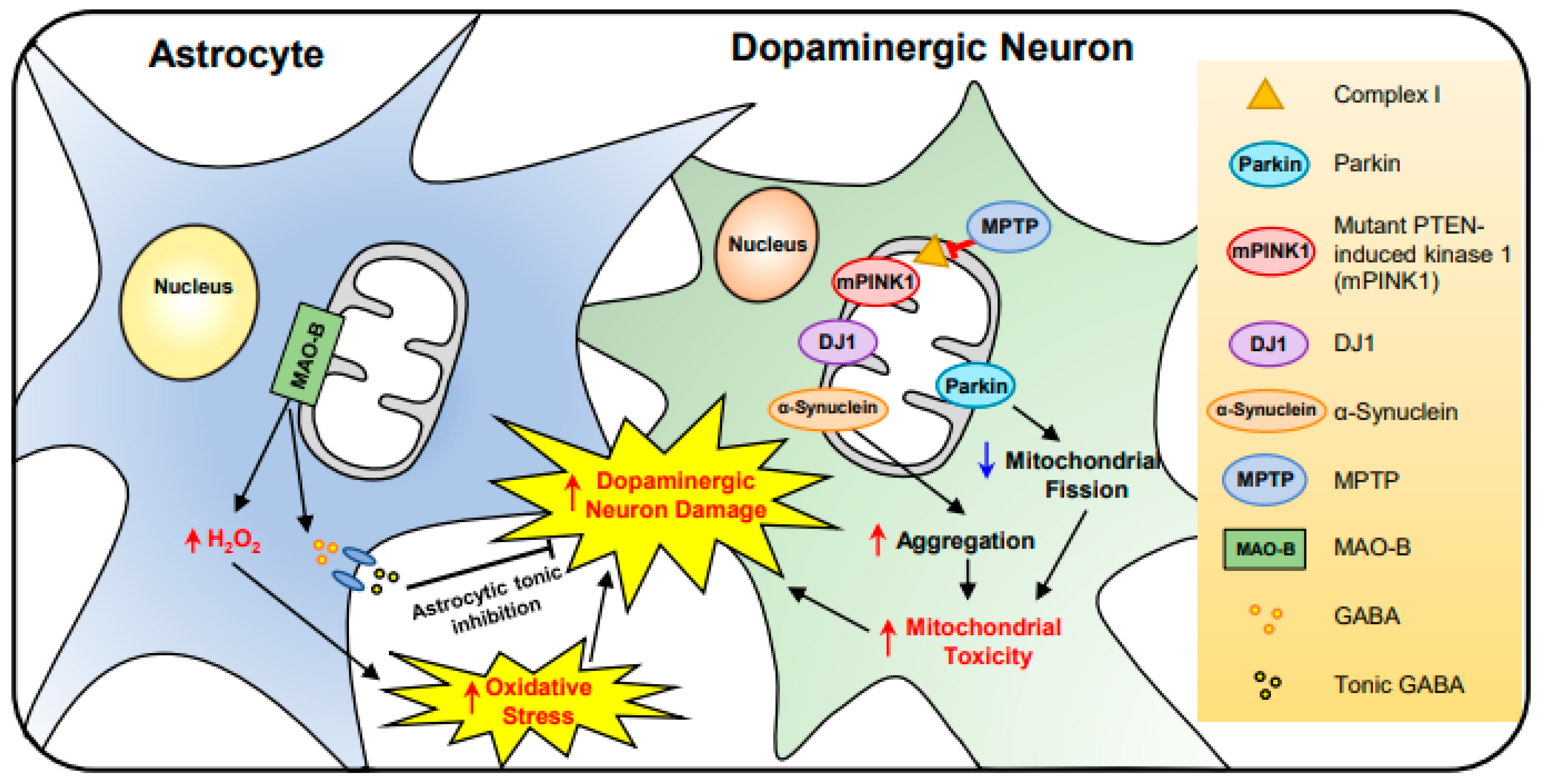
2.4. Parkinson’s Disease (PD)
3. Computational Modeling of Mitochondria in Brain Disorders
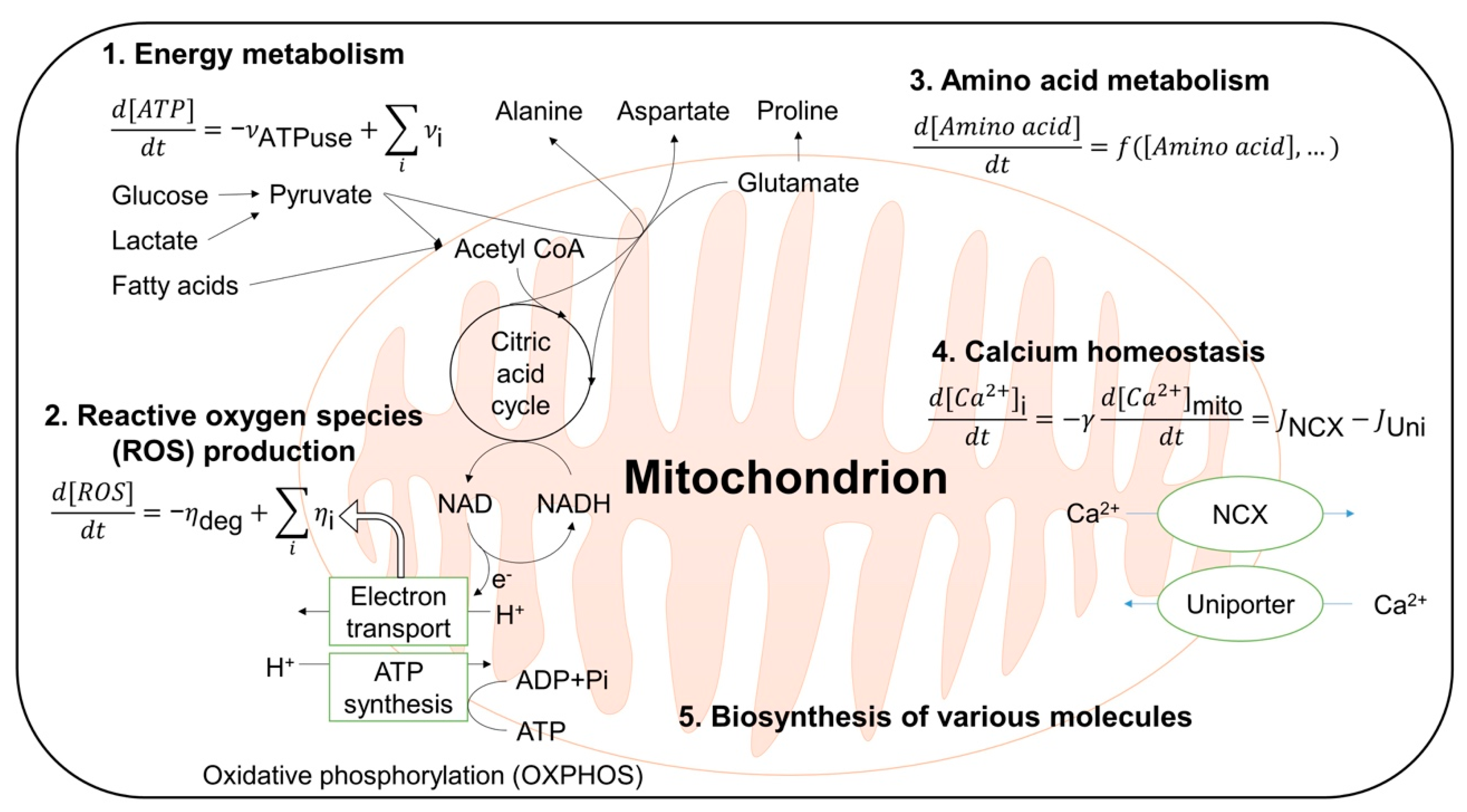
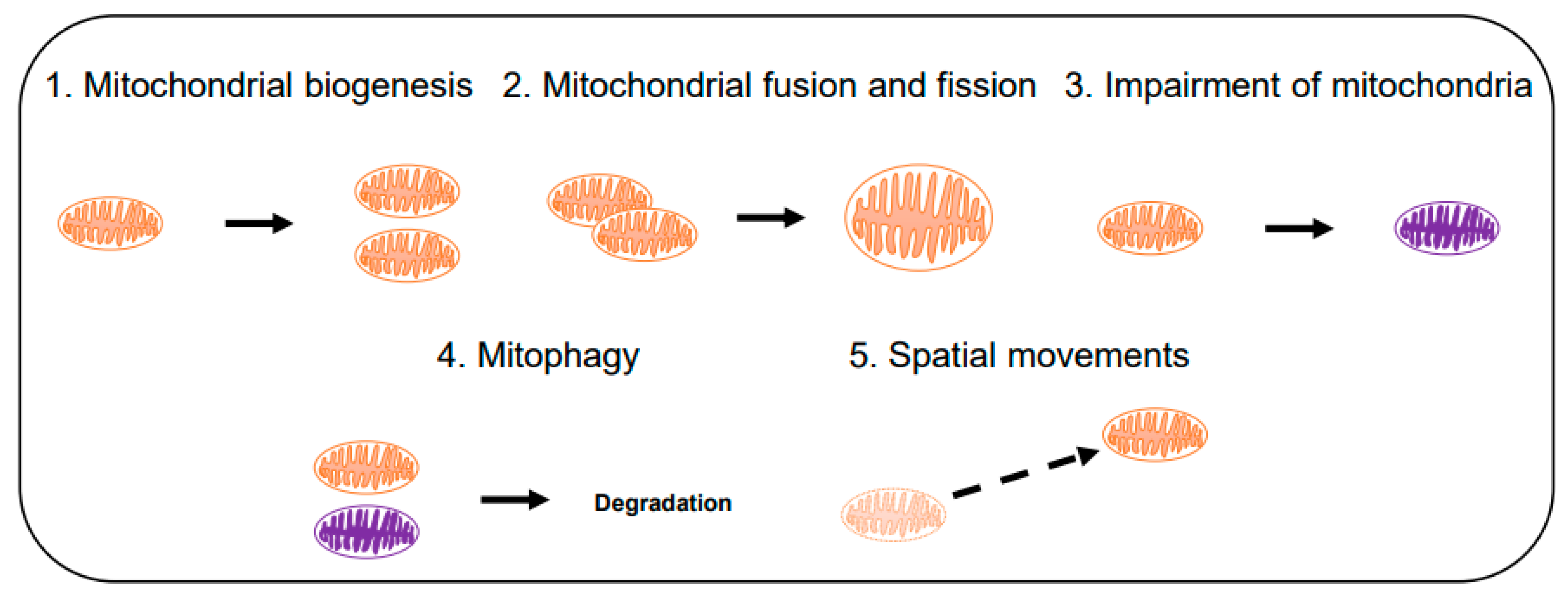
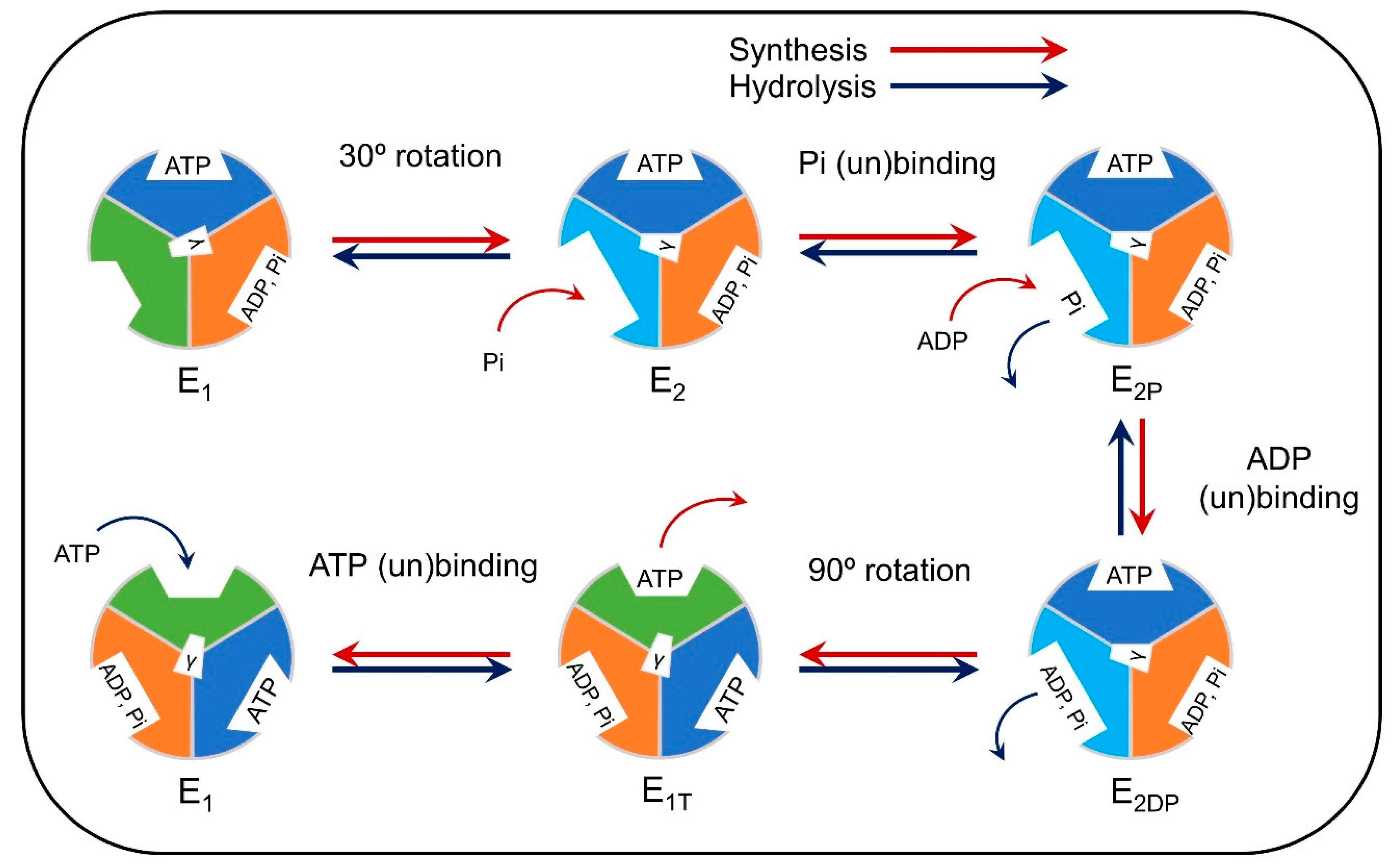
3.1. Computational Modeling of Mitochondria
3.2. Application of Mitochondrial Modeling in Neurodegenerative Diseases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kety, S.S. The General Metabolism of the Brain In Vivo. In Medicine; Springer: New York, NY, USA, 1957; pp. 211–237. [Google Scholar] [CrossRef]
- Sokoloff, L. The Metabolism of the Central Nervous System In Vivo; American Physiolgoical Society: Washington, DC, USA, 1960; Volume 3, pp. 1843–1864. [Google Scholar]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Grossman, L.I.; Schmidt, T.R.; Wildman, D.E.; Goodman, M. Molecular Evolution of Aerobic Energy Metabolism in Primates. Mol. Phylogenet. Evol. 2001, 18, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Langley, B.; Ratan, R.R. Oxidative stress-induced death in the nervous system: Cell cycle dependent or independent? J. Neurosci. Res. 2004, 77, 621–629. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Ohyagi, Y.; Asahara, H.; Chui, D.-H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Aβ42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2004, 19, 255–257. [Google Scholar] [CrossRef]
- Dai, C.-Q.; Luo, T.-T.; Luo, S.-C.; Wang, J.-Q.; Wang, S.-M.; Bai, Y.-H.; Yang, Y.-L.; Wang, Y.-Y. p53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Jembrek, M.J.; Slade, N.; Hof, P.R.; Šimić, G. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.-J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Li, B.; Jia, Z.; Guo, L. Sirtuin 3 mRNA Expression is Downregulated in the Brain Tissues of Alzheimer’s Disease Patients: A Bioinformatic and Data Mining Approach. Med. Sci. Monit. 2020, 26, e923547. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 Is a Subunit of Complex IV of the Mammalian Electron Transport Chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Sze, S.K.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Lee, J.; Boo, J.H.; Ryu, H. The failure of mitochondria leads to neurodegeneration: Do mitochondria need a jump start? Adv. Drug Deliv. Rev. 2009, 61, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.-Y.; Han, S.-H.; Son, S.M.; Hong, H.-S.; Choi, Y.-J.; Byun, J.; Mook-Jung, I. Mitochondria-Specific Accumulation of Amyloid β Induces Mitochondrial Dysfunction Leading to Apoptotic Cell Death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Ota, T.; Matsuoka, Y.; Nomura, Y.; A Smith, M.; Perry, G.; Whitehouse, P.J.; Taniguchi, T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Res. 1998, 780, 260–269. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, S.; Gonnot, F.; Dia, M.; Da Silva, C.C.; Gomez, L.; Sheu, S.-S. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis. 2020, 11, 66. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S.S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and Mitophagy. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, B.; Lee, H.-G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Rui, Y.; Tiwari, P.; Xie, Z.-P.; Zheng, J.Q. Acute Impairment of Mitochondrial Trafficking by beta-Amyloid Peptides in Hippocampal Neurons. J. Neurosci. 2006, 26, 10480–10487. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore: A potential drug target for neurodegeneration. Drug Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Han, P.; Song, M.; Nielsen, M.; Beach, T.G.; Serrano, G.E.; Liang, W.S.; Caselli, R.J.; Shi, J. Amyloid-β Increases Tau by Mediating Sirtuin 3 in Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8592–8601. [Google Scholar] [CrossRef] [PubMed]
- Munguia, M.E.; Govezensky, T.; Martínez, R.; Manoutcharian, K.; Gevorkian, G. Identification of amyloid-beta 1–42 binding protein fragments by screening of a human brain cDNA library. Neurosci. Lett. 2006, 397, 79–82. [Google Scholar] [CrossRef]
- Chen, J.X.; Du Yan, S. Amyloid-β-Induced Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Joh, Y.; Choi, W.-S. Mitochondrial Complex I Inhibition Accelerates Amyloid Toxicity. Dev. Reprod. 2017, 21, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive Oxygen Species: Stuck in the Middle of Neurodegeneration. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S357–S367. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. New Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free. Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Lee, J.; Ryu, H.; Ferrante, R.J.; Morris, S.M., Jr.; Ratan, R.R. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc. Natl. Acad. Sci. USA 2003, 100, 4843–4848. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ryu, H.; Kowall, N. Differential regulation of neuronal and inducible nitric oxide synthase (NOS) in the spinal cord of mutant SOD1 (G93A) ALS mice. Biochem. Biophys. Res. Commun. 2009, 387, 202–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almer, G.; Vukosavic, S.; Romero, N.; Przedborski, S. Inducible Nitric Oxide Synthase Up-Regulation in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 72, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kannagi, M.; Ferrante, R.J.; Kowall, N.W.; Ryu, H. Activation of Ets-2 by oxidative stress induces Bcl-xL expression and accounts for glial survival in amyotrophic lateral sclerosis. FASEB J. 2009, 23, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Ba, M.V.K.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [Green Version]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Demen. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovascular Res. 2013, 10, 222–230. [Google Scholar] [CrossRef]
- Macdonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Parker, W.D.; Boyson, S.J.; Luder, A.S.; Parks, J.K. Evidence for a defect in NADH: Ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology 1990, 40, 1231. [Google Scholar] [CrossRef]
- Arenas, J.; Campos, Y.; Ribacoba, R.; Martín, M.A.; Rubio, J.C.; Ablanedo, P.; Cabello, A. Complex I Defect in muscle from patients with Huntington’s disease. Ann. Neurol. 1998, 43, 397–400. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muquit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Mann, V.M.; Cooper, J.M.; Javoy-Agid, F.; Agid, Y.; Jenner, P.; Schapira, A.H. Mitochondrial function and parental sex effect in Huntington’s disease. Lancet 1990, 336, 749. [Google Scholar] [CrossRef]
- Stahl, W.L.; Swanson, P.D. Biochemical abnormalities in Huntington’s chorea brains. Neurology 1974, 24, 813. [Google Scholar] [CrossRef] [PubMed]
- Kodsi, M.H.; Swerdlow, N.R. Mitochondrial toxin 3-nitropropionic acid produces startle reflex abnormalities and striatal damage in rats that model some features of Huntington’s disease. Neurosci. Lett. 1997, 231, 103–107. [Google Scholar] [CrossRef]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef] [Green Version]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann. Neurol. 1992, 31, 119–130. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef]
- Hansson, O.; Castilho, R.F.; Korhonen, L.; Lindholm, D.; Bates, G.P.; Brundin, P. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington’s disease is dependent on age and CAG repeat length. J. Neurochem. 2001, 78, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Gellerich, F.N.; Gizatullina, Z.; Nguyen, H.P.; Trumbeckaite, S.; Vielhaber, S.; Seppet, E.; Zierz, S.; Landwehrmeyer, B.; Riess, O.; Von Hörsten, S.; et al. Impaired Regulation of Brain Mitochondria by Extramitochondrial Ca2+ in Transgenic Huntington Disease Rats. J. Biol. Chem. 2008, 283, 30715–30724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.M.Y.; Fernandes, H.B.; Zhang, L.Y.J.; Hayden, M.R.; Raymond, L.A. Altered NMDA Receptor Trafficking in a Yeast Artificial Chromosome Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. 2007, 27, 3768–3779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehadeh, J.; Fernandes, H.B.; Mullins, M.M.Z.; Graham, R.K.; Leavitt, B.R.; Hayden, M.R.; Raymond, L.A. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 2006, 21, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.B.; Baimbridge, K.G.; Church, J.; Hayden, M.R.; Raymond, L.A. Mitochondrial Sensitivity and Altered Calcium Handling Underlie Enhanced NMDA-Induced Apoptosis in YAC128 Model of Huntington’s Disease. J. Neurosci. 2007, 27, 13614–13623. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-S.; Tu, H.; Chan, E.Y.; Maximov, A.; Wang, Z.; Wellington, C.L.; Hayden, M.R.; Bezprozvanny, I. Huntingtin and Huntingtin-Associated Protein 1 Influence Neuronal Calcium Signaling Mediated by Inositol-(1,4,5) Triphosphate Receptor Type 1. Neuron 2003, 39, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Lodi, R.; Schapira, A.H.; Manners, D.; Styles, P.; Wood, N.W.; Taylor, D.J.; Warner, T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000, 48, 72–76. [Google Scholar] [CrossRef]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.C.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2010, 121, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S36; discussion S28–S36. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.; Marsden, C. MITOCHONDRIAL COMPLEX I DEFICIENCY IN PARKINSON’S DISEASE. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Gu, M.; Cooper, J.M.; Taanman, J.-W.; Schapira, A.H.V. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann. Neurol. 1998, 44, 177–186. [Google Scholar] [CrossRef]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H.V. Complex I, Iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, M.B. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur. J. Pharmacol. Mol. Pharmacol. 1991, 208, 273–286. [Google Scholar] [CrossRef]
- Jones, N. PINK1 targets dysfunctional mitochondria for autophagy in Parkinson disease. Nat. Rev. Neurol. 2010, 6, 181. [Google Scholar] [CrossRef]
- Moore, D.J.; Zhang, L.; Troncoso, J.; Lee, M.K.; Hattori, N.; Mizuno, Y.; Dawson, T.M.; Dawson, V.L. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 2004, 14, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective Role of the Reaper-Related Serine Protease HtrA2/Omi Revealed by Targeted Deletion in Mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1–19. [Google Scholar] [CrossRef] [Green Version]
- SofroniewHarry, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2009, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Scharr, D.G.; Sieber, B.-A.; Dreyfus, C.F.; Black, I.B. Regional and Cell-Specific Expression of GDNF in Rat Brain. Exp. Neurol. 1993, 124, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal Blood–Brain Barrier Permeability in Parkinson’S Disease. Br. J. Pharmacol. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.Y.; Nam, M.-H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.-J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291.e9. [Google Scholar] [CrossRef]
- Chance, E.M. A computer simulation of oxidative phosphorylation. Comput. Biomed. Res. 1967, 1, 251–264. [Google Scholar] [CrossRef]
- Bohnensack, R. Control of energy transformation in mitochondria. Analysis by a quantitative model. Biochim. Et Biophys. Acta (BBA) Bioenerg. 1981, 634, 203–218. [Google Scholar] [CrossRef]
- Holzhütter, H.-G.; Henke, W.; Dubiel, W.; Gerber, G. A mathematical model to study short-term regulation of mitochondrial energy transduction. Biochim. Et Biophys. Acta (BBA) Bioenerg. 1985, 810, 252–268. [Google Scholar] [CrossRef]
- Korzeniewski, B.; Froncisz, W. An extended dynamic model of oxidative phosphorylation. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 1991, 1060, 210–223. [Google Scholar] [CrossRef]
- Magnus, G.; Keizer, J. Minimal model of beta-cell mitochondrial Ca2+ handling. Am. J. Physiol. Physiol. 1997, 273, C717–C733. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Marbán, E.; Winslow, R.L.; O’Rourke, B. An Integrated Model of Cardiac Mitochondrial Energy Metabolism and Calcium Dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef] [Green Version]
- Beard, D.A. A Biophysical Model of the Mitochondrial Respiratory System and Oxidative Phosphorylation. PLoS Comput. Biol. 2005, 1, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Yang, F.; Vinnakota, K.C.; Beard, D.A. Computer Modeling of Mitochondrial Tricarboxylic Acid Cycle, Oxidative Phosphorylation, Metabolite Transport, and Electrophysiology. J. Biol. Chem. 2007, 282, 24525–24537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazil, J.N.; Buzzard, G.T.; Rundell, A.E. Modeling Mitochondrial Bioenergetics with Integrated Volume Dynamics. PLoS Comput. Biol. 2010, 6, e1000632. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A. Computational Modeling of Mitochondrial Function. Toxic. Assess. 2011, 810, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V. Thermodynamics and control of proton motive free-energy transduction. Biomed. Biochim. Acta 1985, 44, 929–941. [Google Scholar]
- Tager, J.; Wanders, R.; Groen, A.; Kunz, W.; Bohnensack, R.; Küster, U.; Letko, G.; Böhme, G.; Duszynski, J.; Wojtczak, L. Control of mitochondrial respiration. FEBS Lett. 1983, 151, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V.; Lolkema, J.S.; Otto, R.; Hellingwerf, K.J. Thermodynamics of growth non-equilibrium thermodynamics of bacterial growth the phenomenological and the Mosaic approach. Biochim. Et Biophys. Acta (BBA) Rev. Bioenerg. 1982, 683, 181–220. [Google Scholar] [CrossRef]
- Stucki, J.W. The Optimal Efficiency and the Economic Degrees of Coupling of Oxidative Phosphorylation. JBIC J. Biol. Inorg. Chem. 1980, 109, 269–283. [Google Scholar] [CrossRef]
- Pietrobon, D.; Zoratti, M.; Azzone, G.F.; Caplan, S.R. Intrinsic uncoupling of mitochondrial proton pumps. 2. Modeling studies. Biochemistry 1986, 25, 767–775. [Google Scholar] [CrossRef]
- Christensen, B.; Nielsen, J. Metabolic network analysis. A powerful tool in metabolic engineering. Adv. Biochem. Eng. 2000, 66, 209–231. [Google Scholar]
- Savinell, J.M.; Palsson, B.O. Network analysis of intermediary metabolism using linear optimization. I. Development of mathematical formalism. J. Theor. Biol. 1992, 154, 421–454. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, J.C.; Aon, M.A. Fluxes of carbon, phosphorylation, and redox intermediates during growth ofsaccharomyces cerevisiae on different carbon sources. Biotechnol. Bioeng. 1995, 47, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Mathematical models in molecular and cellular biology. Acta Appl. Math. 1985, 4, 267–268. [CrossRef]
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A Mitochondrial Oscillator Dependent on Reactive Oxygen Species. Biophys. J. 2004, 87, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.D.; Greenstein, J.L.; O’Rourke, B.; Winslow, R.L. An Integrated Mitochondrial ROS Production and Scavenging Model: Implications for Heart Failure. Biophys. J. 2013, 105, 2832–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L.; Kowald, A. The free-radical theory of ageing–older, wiser and still alive: Modelling positional effects of the primary targets of ROS reveals new support. BioEssays 2012, 34, 692–700. [Google Scholar] [CrossRef]
- Pezze, P.D.; Nelson, G.; Otten, E.G.; Korolchuk, V.I.; Kirkwood, T.B.L.; Von Zglinicki, T.; Shanley, D.P. Dynamic Modelling of Pathways to Cellular Senescence Reveals Strategies for Targeted Interventions. PLoS Comput. Biol. 2014, 10, e1003728. [Google Scholar] [CrossRef] [Green Version]
- De Brito, P.M.; Antunes, F. Estimation of kinetic parameters related to biochemical interactions between hydrogen peroxide and signal transduction proteins. Front. Chem. 2014, 2, 82. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, A.; Haanstra, J.R.; Engqvist, M.K.; Gerding, A.; Bakker, B.M.; Klingmüller, U.; Teusink, B.; Nielsen, J. Quantitative analysis of amino acid metabolism in liver cancer links glutamate excretion to nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10294–10304. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Marhl, M.; Höfer, T.T. Modelling of simple and complex calcium oscillations. From single-cell responses to intercellular signalling. JBIC J. Biol. Inorg. Chem. 2002, 269, 1333–1355. [Google Scholar] [CrossRef]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons inpink1−/−zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.D.; Lindahl, P.A. A mathematical model of iron import and trafficking in wild-type and Mrs3/4ΔΔ yeast cells. BMC Syst. Biol. 2019, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Melser, S.; Lavie, J.; Bénard, G. Mitochondrial degradation and energy metabolism. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.L.; Chourasia, A.H.; MacLeod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Kowald, A.; Kirkwood, T. Mitochondrial mutations, cellular instability and ageing: Modelling the population dynamics of mitochondria. Mutat. Res. 1993, 295, 93–103. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T. A network theory of ageing: The interactions of defective mitochondria, aberrant proteins, free radicals and scavengers in the ageing process. Mutat. Res. 1996, 316, 209–236. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B. Accumulation of Defective Mitochondria through Delayed Degradation of Damaged Organelles and Its Possible Role in the Ageing of Post-mitotic and Dividing Cells. J. Theor. Biol. 2000, 202, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B.L. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc. Natl. Acad. Sci. USA 2011, 108, 10237–10242. [Google Scholar] [CrossRef] [Green Version]
- Mouli, P.K.; Twig, G.; Shirihai, O.S. Frequency and Selectivity of Mitochondrial Fusion Are Key to Its Quality Maintenance Function. Biophys. J. 2009, 96, 3509–3518. [Google Scholar] [CrossRef] [Green Version]
- Tam, Z.Y.; Gruber, J.; Halliwell, B.; Gunawan, R. Mathematical Modeling of the Role of Mitochondrial Fusion and Fission in Mitochondrial DNA Maintenance. PLoS ONE 2013, 8, e76230. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.K.; Shirihai, O.; Huang, K.C. Optimal Dynamics for Quality Control in Spatially Distributed Mitochondrial Networks. PLoS Comput. Biol. 2013, 9, e1003108. [Google Scholar] [CrossRef] [Green Version]
- Dalmasso, G.; Zapata, P.A.M.; Brady, N.R.; Hamacher-Brady, A. Agent-Based Modeling of Mitochondria Links Sub-Cellular Dynamics to Cellular Homeostasis and Heterogeneity. PLoS ONE 2017, 12, e0168198. [Google Scholar] [CrossRef] [Green Version]
- Kornick, K.; Bogner, B.; Sutter, L.; Das, M. Population Dynamics of Mitochondria in Cells: A Minimal Mathematical Model. Front. Phys. 2019, 7. [Google Scholar] [CrossRef]
- Boyer, P.D. The Atp Synthase—A Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Terni, B.; Boada, J.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Mitochondrial ATP-Synthase in the Entorhinal Cortex Is a Target of Oxidative Stress at Stages I/II of Alzheimer’s Disease Pathology. Brain Pathol. 2010, 20, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Xi, Y.; Gao, M.; Li, Z.; Xu, C.; Fan, S.; He, W. Gene Expression Profiles of Entorhinal Cortex in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dementiasr 2014, 29, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, W.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Tian, J.; Guo, L.; Du, H. Caspase inhibition rescues F1Fo ATP synthase dysfunction-mediated dendritic spine elimination. Sci. Rep. 2020, 10, 17589. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2011, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Tebbenkamp, A.T.N.; Varela, L.; Choi, J.; Paredes, M.I.; Giani, A.M.; Song, J.E.; Sestan-Pesa, M.; Franjic, D.; Sousa, A.M.M.; Liu, Z.-W.; et al. The 7q11.23 Protein DNAJC30 Interacts with ATP Synthase and Links Mitochondria to Brain Development. Cell 2018, 175, 1088–1104.e23. [Google Scholar] [CrossRef] [Green Version]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, M.; Carelli, V.; Solaini, G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Pietrobon, D.; Caplan, S.R. Flow-force relationships for a six-state proton pump model: Intrinsic uncoupling, kinetic equivalence of input and output forces, and domain of approximate linearity. Biochemistry 1985, 24, 5764–5776. [Google Scholar] [CrossRef]
- Gao, Y.Q.; Yang, W.; Karplus, M. A Structure-Based Model for the Synthesis and Hydrolysis of ATP by F1-ATPase. Cell 2005, 123, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Boyer, P.D. The binding change mechanism for ATP synthase—Some probabilities and possibilities. Biochim. Et Biophys. Acta (BBA) Bioenerg. 1993, 1140, 215–250. [Google Scholar] [CrossRef]
- Noji, H.; Yasuda, R.; Yoshida, M.; Kinosita, K., Jr. Direct observation of the rotation of F1-ATPase. Nat. Cell Biol. 1997, 386, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P.; Leslie, A.G.W.; Lutter, R.; Walker, J.E. Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria. Nat. Cell Biol. 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Menz, R.; Walker, J.E.; Leslie, A.G. Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: Implications for the mechanism of rotary catalysis. Cell 2001, 106, 331–341. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.W.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar] [CrossRef] [Green Version]
- Dautant, A.; Velours, J.; Giraud, M.-F. Crystal Structure of the Mg·ADP-inhibited State of the Yeast F1c10-ATP Synthase. J. Biol. Chem. 2010, 285, 29502–29510. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Flynn, T.C.; Cui, Q.; Leslie, A.G.W.; E Walker, J.; Karplus, M. A dynamic analysis of the rotation mechanism for conformational change in F(1)-ATPase. Structure 2002, 10, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Bockmann, R.A.; Grubmuller, H. Nanoseconds molecular dynamics simulation of primary mechanical energy transfer steps in F1-ATP synthase. Nat. Genet. 2002, 9, 198–202. [Google Scholar] [CrossRef]
- Symersky, J.; Pagadala, V.; Osowski, D.; Krah, A.; Meier, T.; Faraldo-Gómez, J.D.; Mueller, D.M. Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat. Struct. Mol. Biol. 2012, 19, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Kubo, S.; Niina, T.; Takada, S. Molecular dynamics simulation of proton-transfer coupled rotations in ATP synthase FO motor. Sci. Rep. 2020, 10, 8225. [Google Scholar] [CrossRef]
- Zhou, W.; Marinelli, F.; Nief, C.; Faraldo-Gómez, J.D. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. eLife 2017, 6, 10580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; E Tosatto, S.C.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkian, W.; König, M.; Wassenaar, T.A.; Marrink, S.J. Backmapping triangulated surfaces to coarse-grained membrane models. Nat. Commun. 2020, 11, 2296. [Google Scholar] [CrossRef] [PubMed]
- Raichur, A.; Vali, S.; Gorin, F. Dynamic modeling of alpha-synuclein aggregation for the sporadic and genetic forms of Parkinson’s disease. Neuroscience 2006, 142, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; Wellstead, P. Dynamic modelling of protein and oxidative metabolisms simulates the pathogenesis of Parkinson’s disease. IET Syst. Biol. 2012, 6, 65–72. [Google Scholar] [CrossRef]
- Cloutier, M.; Middleton, R.; Wellstead, P. Feedback motif for the pathogenesis of Parkinson’s disease. IET Syst. Biol. 2012, 6, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, S.; Chelliah, V.; Chen, C.; Van Der Graaf, P.H. Mathematical Biology Models of Parkinson’s Disease. CPT 2019, 8, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Friedman, A. Mathematical model on Alzheimer’s disease. BMC Syst. Biol. 2016, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, B.; Chong, K.H.; Zheng, J. Composite mathematical modeling of calcium signaling behind neuronal cell death in Alzheimer’s disease. BMC Syst. Biol. 2018, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Kim, J.; Choi, M.Y. Autophagy mediates phase transitions from cell death to life. Heliyon 2015, 1, e00027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Kim, S.H.; Choi, M.Y. Computational modeling of the effects of autophagy on amyloid-β peptide levels. Theor. Biol. Med. Model. 2020, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Kim, J.; Choi, M.Y. Quantitative indices of autophagy activity from minimal models. Theor. Biol. Med. Model. 2014, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Kim, J.; Choi, M. Computer simulations unveil the dynamics of autophagy and its implications for the cellular qual-ity control. J. Biol. Syst. 2014, 22, 659–675. [Google Scholar] [CrossRef]
- Han, K.; Kwon, H.W.; Kang, H.; Kim, J.; Lee, M.S.; Choi, M.Y. Dynamics of macroautophagy: Modeling and oscillatory be-havior. Phys. A 2012. [Google Scholar] [CrossRef]
- Barbeito, L.; Pehar, M.; Cassina, P.; Vargas, M.R.; Peluffo, H.; Viera, L.; Estévez, A.G.; Beckman, J.S. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Rev. 2004, 47, 263–274. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2− production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229. https://doi.org/10.3390/antiox10020229
Woo J, Cho H, Seol Y, Kim SH, Park C, Yousefian-Jazi A, Hyeon SJ, Lee J, Ryu H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants. 2021; 10(2):229. https://doi.org/10.3390/antiox10020229
Chicago/Turabian StyleWoo, JunHyuk, Hyesun Cho, YunHee Seol, Soon Ho Kim, Chanhyeok Park, Ali Yousefian-Jazi, Seung Jae Hyeon, Junghee Lee, and Hoon Ryu. 2021. "Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models" Antioxidants 10, no. 2: 229. https://doi.org/10.3390/antiox10020229
APA StyleWoo, J., Cho, H., Seol, Y., Kim, S. H., Park, C., Yousefian-Jazi, A., Hyeon, S. J., Lee, J., & Ryu, H. (2021). Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants, 10(2), 229. https://doi.org/10.3390/antiox10020229