
Disruption of Endoplasmic Reticulum and ROS Production in Human Ovarian Cancer by Campesterol



Abstract
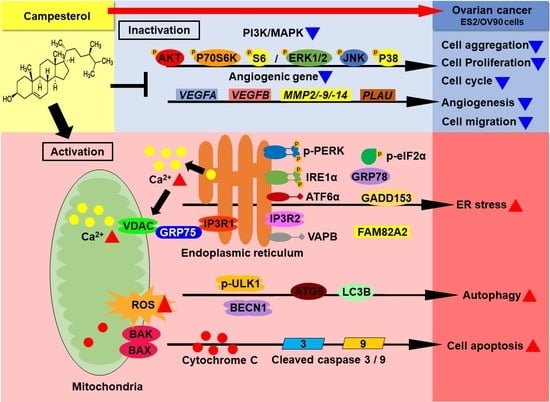
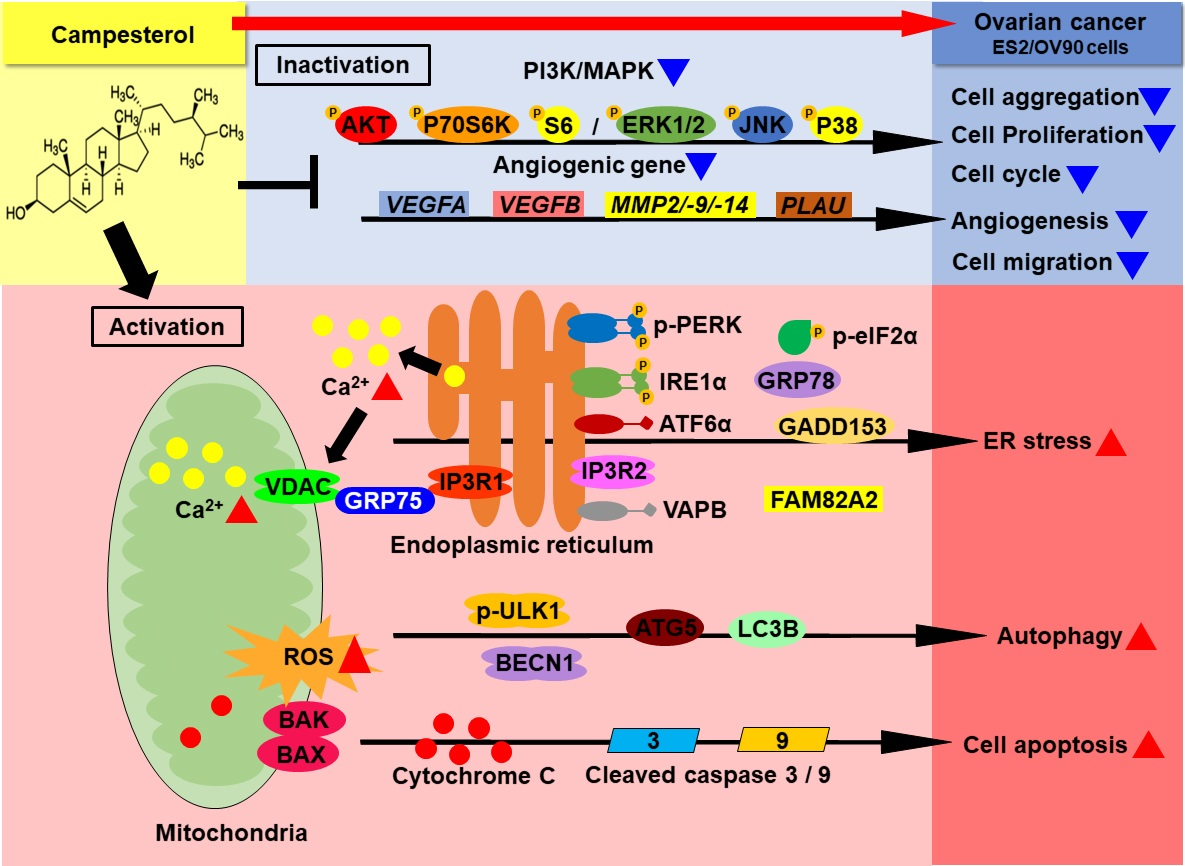
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. 3D Cell Culture
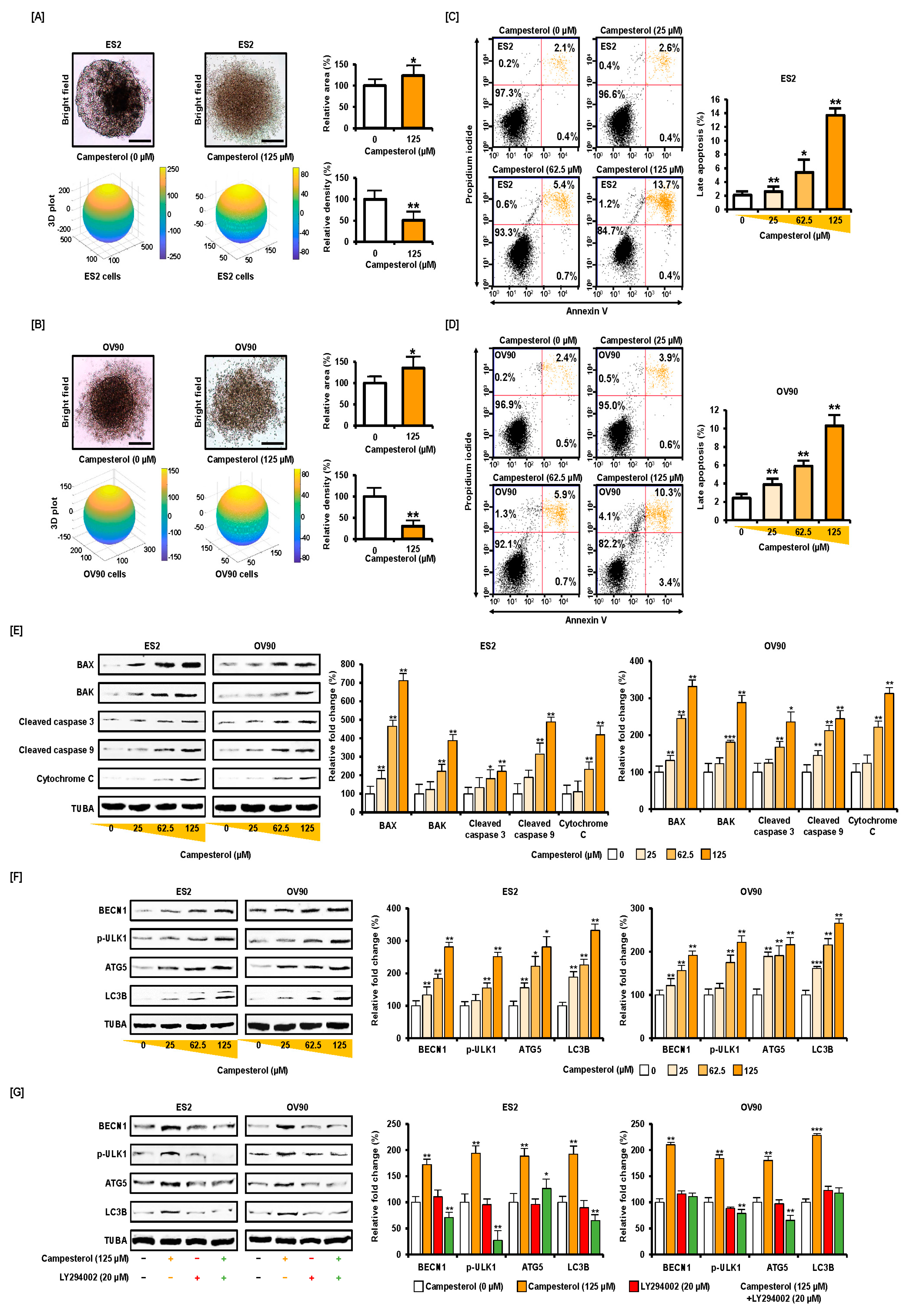
2.4. Apoptosis Assay
2.5. Western Blot Analysis
2.6. JC-1 Staining
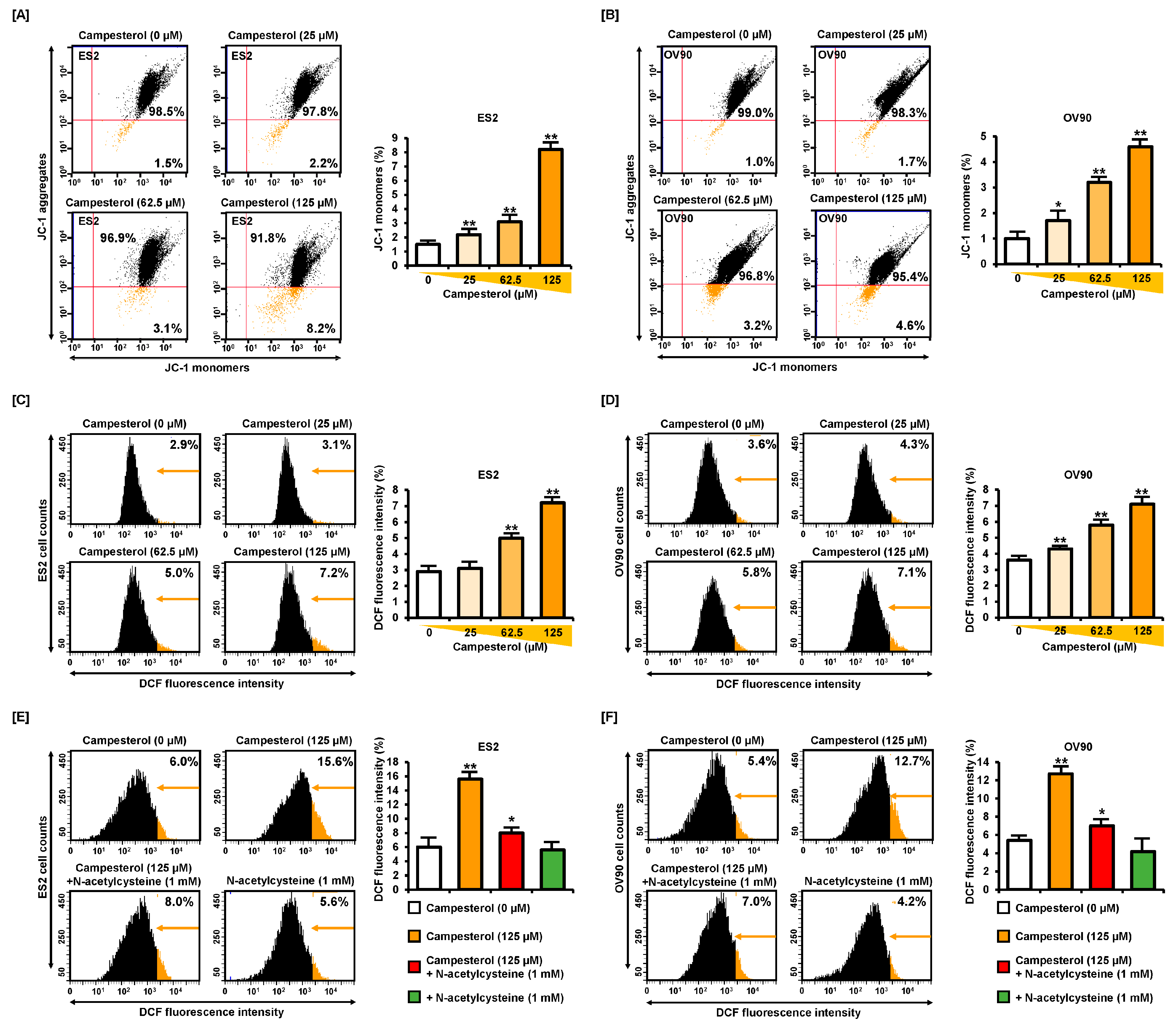
2.7. ROS Production
2.8. Evaluation of Cytosolic Calcium Levels
2.9. Evaluation of Mitochondrial Calcium Levels
2.10. Cell Proliferation
2.11. Cell Cycle
2.12. Migration
2.13. Quantitative Real Time PCR
2.14. Statistical Analysis
3. Results
3.1. Activation of Cell Death and Restriction of Cell Aggregation by Campesterol in Human Ovarian Cancer Cells
3.2. Alterations in the Mitochondrial Membrane Potential (MMP) and ROS Levels by Campesterol
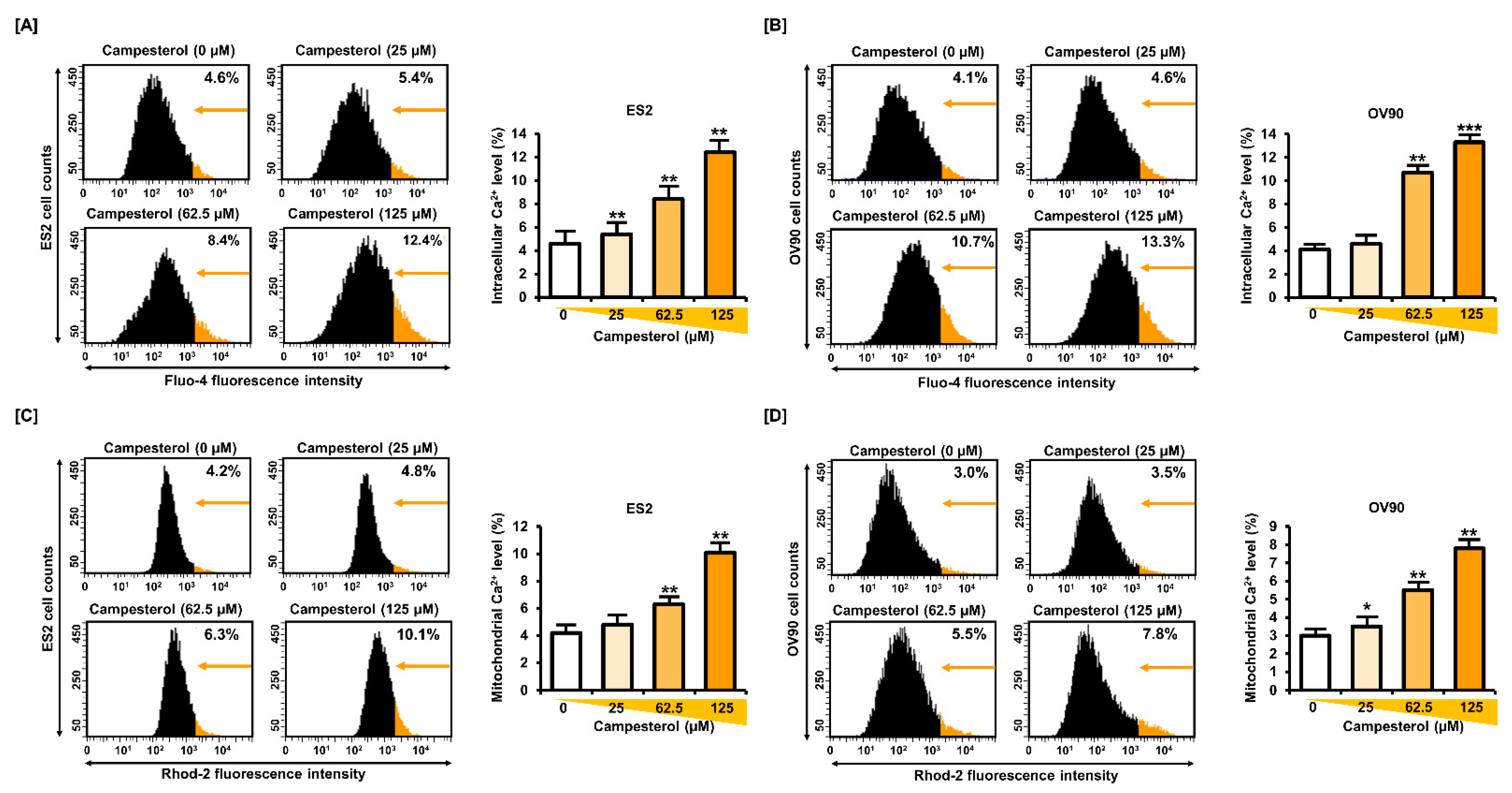
3.3. Upregulation of the Cellular and Mitochondrial Calcium Concentrations by Campesterol
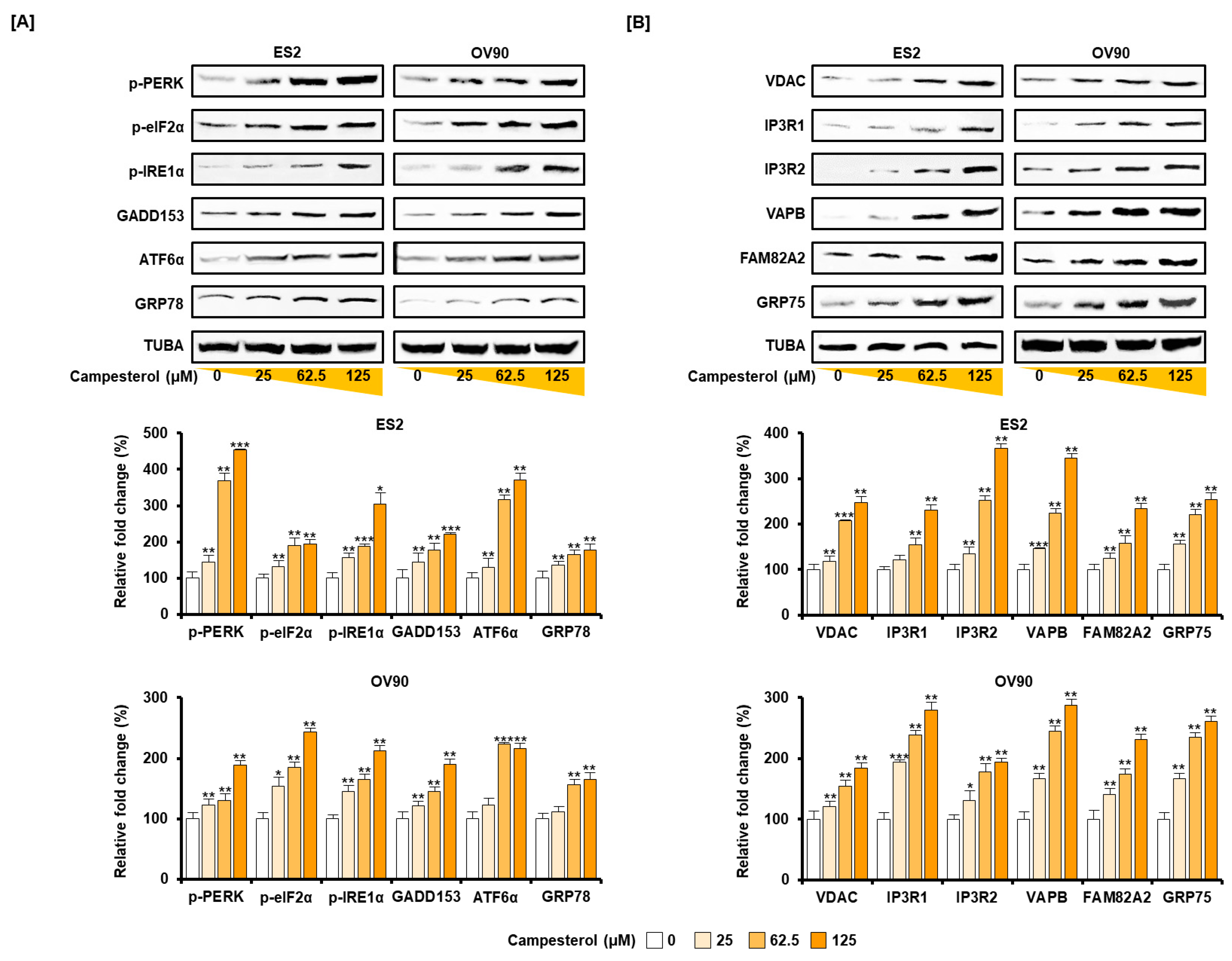
3.4. Activation of ER Stress and the ER–Mitochondrial Axis by Campesterol in the ES2 and OV90 Cells
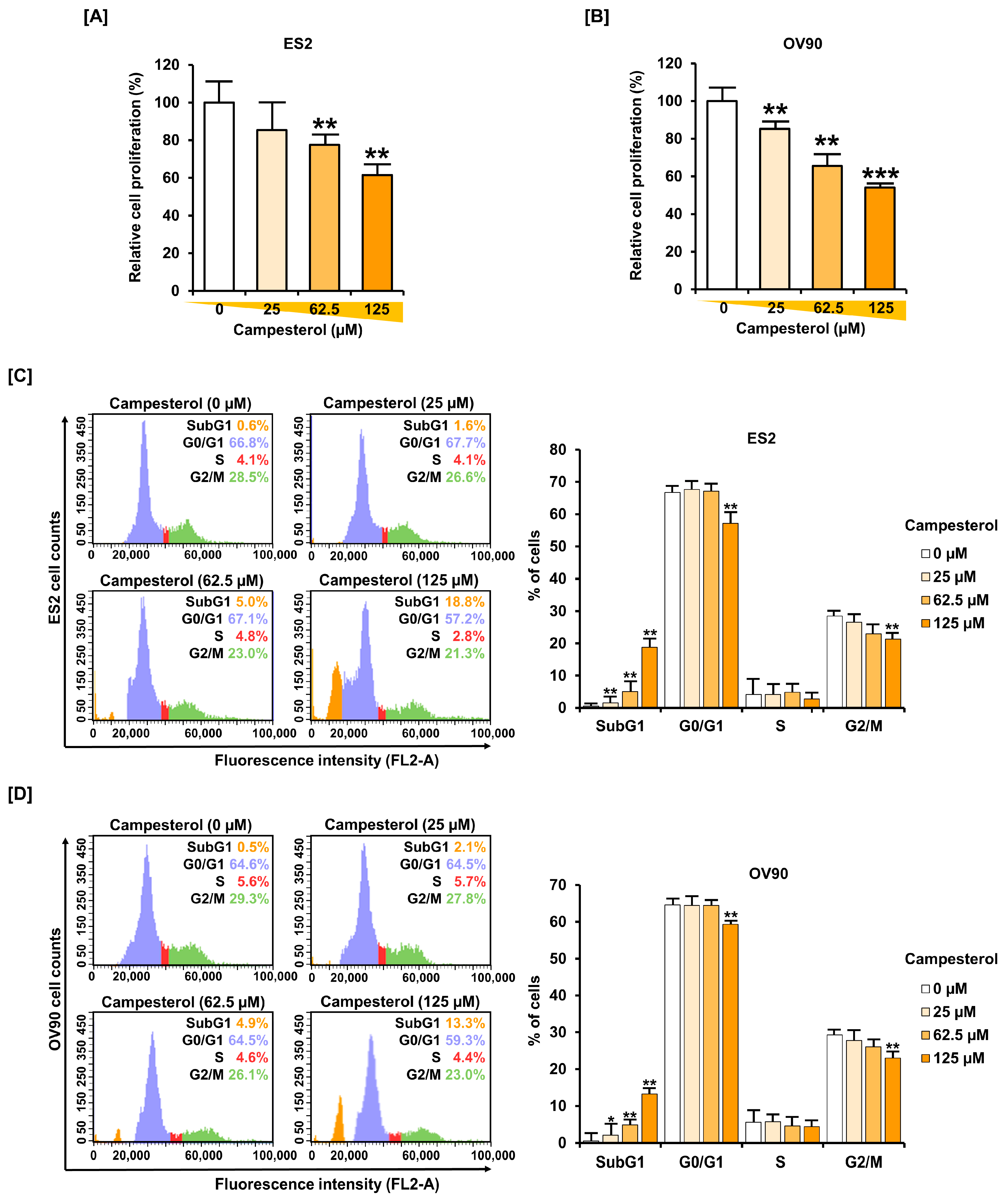
3.5. Inhibition of Cell Proliferation and the Cell Cycle by Campesterol
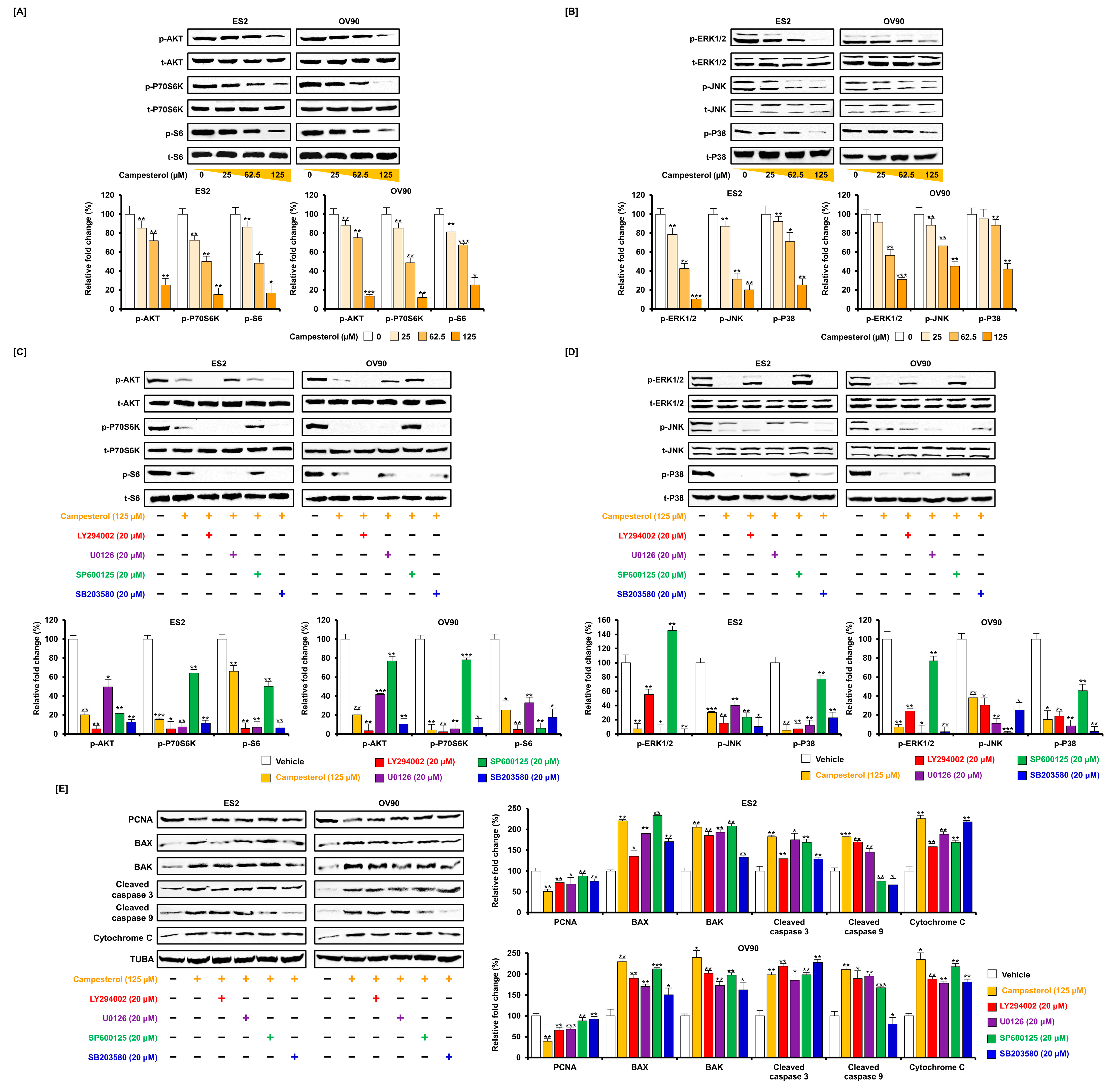
3.6. Regulation of Signaling by Campesterol in the Ovarian Cancer Cells
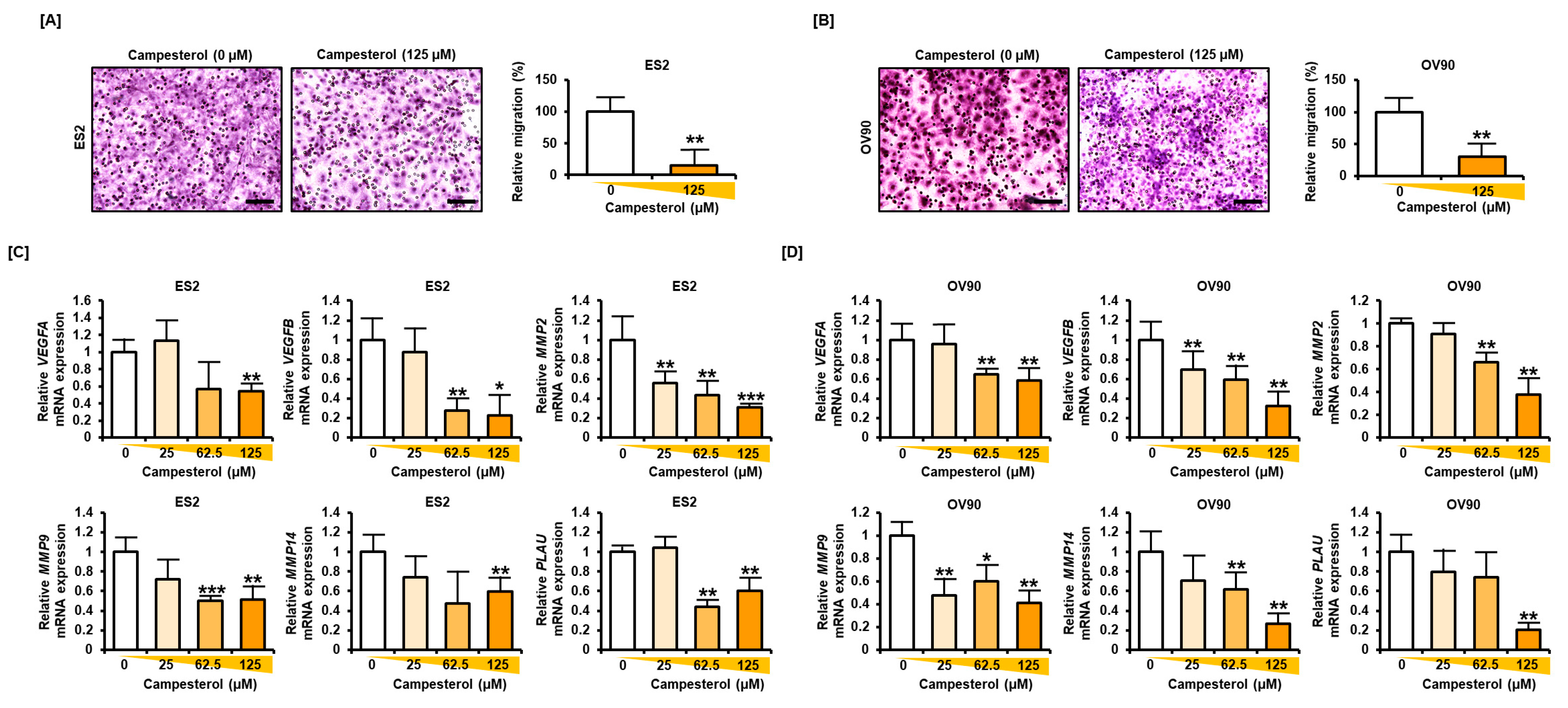
3.7. Reductions in the Migration and Expression of Angiogenic Genes in the Ovarian Cancer Cells Treated with Campesterol
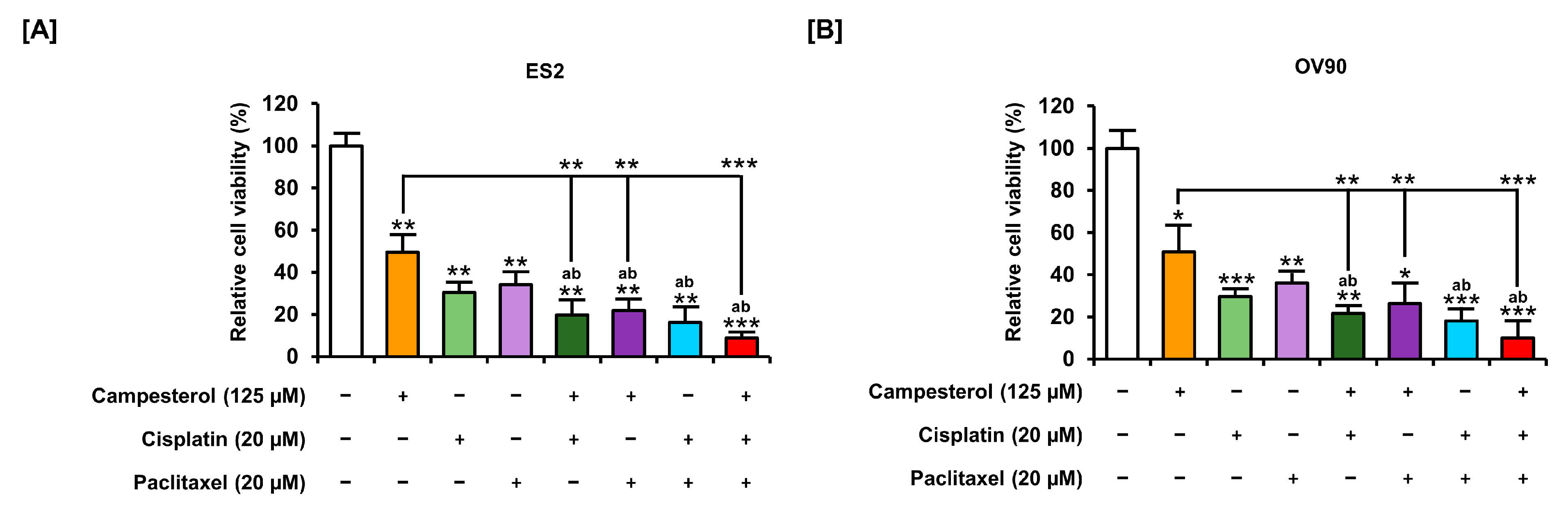
3.8. Enhancements in the Anticancer Effects of Existing Drugs by Campesterol
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Genser, B.; Silbernagel, G.; De Backer, G.; Bruckert, E.; Carmena, R.; Chapman, M.J.; Deanfield, J.; Descamps, O.S.; Rietzschel, E.R.; Dias, K.C.; et al. Plant sterols and cardiovascular disease: A systematic review and meta-analysis†. Eur. Hear. J. 2012, 33, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Lea, L.; Hepburn, P. Safety evaluation of phytosterol-esters. Part 9: Results of a European post-launch monitoring programme. Food Chem. Toxicol. 2006, 44, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, K.; Pullen, M.L.; Topping, J.F. Importance of plant sterols in pattern formation and hormone signalling. Trends Plant. Sci. 2003, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, P. Biosynthesis and Accumulation of Sterols. Annu. Rev. Plant. Biol. 2004, 55, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Wang, Q.; Chong, K.; Wang, F.; Wang, L.; Bai, M.; Jia, C. The brassinosteroid signal transduction pathway. Cell Res. 2006, 16, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Mongrand, S.; Morel, J.; Laroche, J.; Claverol, S.; Carde, J.-P.; Hartmann, M.-A.; Bonneu, M.; Simon-Plas, F.; Lessire, R.; Bessoule, J.-J. Lipid Rafts in Higher Plant Cells. J. Biol. Chem. 2004, 279, 36277–36286. [Google Scholar] [CrossRef] [Green Version]
- Laloi, M.; Perret, A.-M.; Chatre, L.; Melser, S.; Cantrel, C.; Vaultier, M.-N.; Zachowski, A.; Bathany, K.; Schmitter, J.-M.; Vallet, M.; et al. Insights into the Role of Specific Lipids in the Formation and Delivery of Lipid Microdomains to the Plasma Membrane of Plant Cells. Plant. Physiol. 2006, 143, 461–472. [Google Scholar] [CrossRef] [Green Version]
- McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Graham, S. Risk of Human Ovarian Cancer Is Related to Dietary Intake of Selected Nutrients, Phytochemicals and Food Groups. J. Nutr. 2003, 133, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Segura, R.; Javierre, C.; Lizarraga, M.A.; Ros, E. Other relevant components of nuts: Phytosterols, folate and minerals. Br. J. Nutr. 2006, 96, S36–S44. [Google Scholar] [CrossRef] [Green Version]
- Calpe-Berdiel, L.; Escolà-Gil, J.C.; Blanco-Vaca, F. New insights into the molecular actions of plant sterols and stanols in cholesterol metabolism. Atherosclerosis 2009, 203, 18–31. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, Y.; Kenny, O.; O’Connell, N.M.; Maguire, A.R.; McCarthy, F.O.; O’Brien, N.M. Synthesis and assessment of the relative toxicity of the oxidised derivatives of campesterol and dihydrobrassicasterol in U937 and HepG2 cells. Biochimie 2013, 95, 496–503. [Google Scholar] [CrossRef]
- Chuu, C.-P.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Modulation of liver X receptor signaling as novel therapy for prostate cancer. J. Biomed. Sci. 2007, 14, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.B.; Fink, C.S.; Williams, H.; Kim, U. In vitro and in vivo (SCID mice) effects of phytosterols on the growth and dissemination of human prostate cancer PC-3 cells. Eur. J. Cancer Prev. 2001, 10, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhao, X.; Xu, J.; Li, C.; Yu, Y.; Wang, W.; Zhu, L. The Protective Effect of Dietary Phytosterols on Cancer Risk: A Systematic Meta-Analysis. J. Oncol. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggan, M.A.; Anderson, W.F.; Altekruse, S.; Penberthy, L.; Sherman, M.E. The Surveillance, Epidemiology, and End Results (SEER) Program and Pathology. Am. J. Surg. Pathol. 2016, 40, e94–e102. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Mph, K.D.M.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Grasso, D.; Bouzin, C.; Brusa, D.; Rossignol, R.; Sonveaux, P. Mitochondria Participate in Chemoresistance to Cisplatin in Human Ovarian Cancer Cells. Mol. Cancer Res. 2020, 18, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Nautiyal, J.; Xiao, Y.; Curry, E.; Sun, M.; Zanini, E.; Paige, A.J.; Gabra, H. WWOX sensitises ovarian cancer cells to paclitaxel via modulation of the ER stress response. Cell Death Dis. 2017, 8, e2955. [Google Scholar] [CrossRef]
- Bae, H.; Song, G.; Lee, J.-Y.; Hong, T.; Chang, M.-J.; Lim, W. Laminarin-Derived from Brown Algae Suppresses the Growth of Ovarian Cancer Cells via Mitochondrial Dysfunction and ER Stress. Mar. Drugs 2020, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.; Lee, J.-Y.; Yang, C.; Song, G.; Lim, W. Fucoidan Derived from Fucus vesiculosus Inhibits the Development of Human Ovarian Cancer via the Disturbance of Calcium Homeostasis, Endoplasmic Reticulum Stress, and Angiogenesis. Mar. Drugs 2020, 18, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.B.; Downie, A.C.; Fink, C.S. Inhibition of growth and stimulation of apoptosis by beta-sitosterol treatment of MDA-MB-231 human breast cancer cells in culture. Int. J. Mol. Med. 2000, 5, 541–546. [Google Scholar] [CrossRef]
- Belayachi, L.; Aceves-Luquero, C.; Merghoub, N.; De Mattos, S.F.; Amzazi, S.; Villalonga, P.; Bakri, Y. Induction of cell cycle arrest and apoptosis by ormenis eriolepis a morrocan endemic plant in various human cancer cell lines. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 356–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.-F.; Huang, K.-H.; Lu, K.-J.; Chiou, S.-J.; Yen, J.-H.; Chang, C.-C.; Houng, J.-Y. Typhonium blumei extract inhibits proliferation of human lung adenocarcinoma A549 cells via induction of cell cycle arrest and apoptosis. J. Ethnopharmacol. 2011, 135, 492–500. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, C.; Zu, Y.; Song, Z.; Zhang, B.; Meng, X.; Qiu, W.; Zhang, L. SFE-CO2 Extract from Typhonium giganteum Engl. Tubers, Induces Apoptosis in Human Hepatoma SMMC-7721 Cells Involvement of a ROS-Mediated Mitochondrial Pathway. Molecules 2011, 16, 8228–8243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2003, 3, 35–40. [Google Scholar] [CrossRef]
- Tirmenstein, M.A.; Hu, C.X.; Gales, T.L.; Maleeff, B.E.; Narayanan, P.K.; Kurali, E.; Hart, T.K.; Thomas, H.C.; Schwartz, L.W. Effects of troglitazone on HepG2 viability and mitochondrial function. Toxicol. Sci. 2002, 69, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Mann, S.; Hammarback, J. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem. 1994, 269, 11492–11497. [Google Scholar] [CrossRef]
- Lang, T.; Schaeffeler, E.; Bernreuther, D.; Bredschneider, M.; Wolf, D.H.; Thumm, M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998, 17, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Dang, Y.; Su, W.; Liu, C.; Ma, H.; Shan, Y.; Pei, Y.; Wan, B.; Guo, J.; Yu, L. Molecular cloning and characterization of rat LC3A and LC3B—Two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 2006, 339, 437–442. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nat. Cell Biol. 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Chan, E.Y. Regulation and Function of Uncoordinated-51 Like Kinase Proteins. Antioxid. Redox Signal. 2012, 17, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.-I.; Natsume, T.; Guan, J.-L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef] [Green Version]
- Knickle, A.; Rasmussen, A.; Hoskin, D.W. Myricetin induces apoptosis mediated by oxidative stress in 4T1 and E0771 mammary cancer cells. Mol. Cell. Toxicol. 2020, 16, 283–289. [Google Scholar] [CrossRef]
- Larman, M.; Lucy, J. Meeting Report. Mol. Membr. Biol. 1999, 16, 313–316. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Marelli, M.M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [Green Version]
- Mandl, J.; Mészáros, T.; Bánhegyi, G.; Hunyady, L.; Csala, M. Endoplasmic reticulum: Nutrient sensor in physiology and pathology. Trends Endocrinol. Metab. 2009, 20, 194–201. [Google Scholar] [CrossRef]
- Gao, A.W.; Cantó, C.; Houtkooper, R.H. Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol. Med. 2014, 6, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nat. Cell Biol. 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Xie, Q.; Su, J.; Jiao, B.; Shen, L.; Ma, L.; Qu, X.; Yu, C.; Jiang, X.; Xu, Y.; Sun, L. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 2016, 49, 2507–2519. [Google Scholar] [CrossRef] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Kazłowska, K.; Lin, H.-T.V.; Chang, S.-H.; Tsai, G.-J. In VitroandIn VivoAnticancer Effects of Sterol Fraction from Red AlgaePorphyra dentata. Evid.-Based Complement. Altern. Med. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Tribioli, C.; Prosperi, E. Targeting Proliferating Cell Nuclear Antigen (PCNA) as an Effective Strategy to Inhibit Tumor Cell Proliferation. Curr. Cancer Drug Targets 2020, 20, 240–252. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhan, Y.; Xu, J.; Wang, Y.; Sun, M.; Chen, J.; Liang, T.; Wu, L.; Xu, K. β-Sitosterol Reverses Multidrug Resistance via BCRP Suppression by Inhibiting the p53–MDM2 Interaction in Colorectal Cancer. J. Agric. Food Chem. 2020, 68, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmings, E.; Mullany, S.; Chang, Z.; Landen, J.C.N.; Linder, S.; Bazzaro, M. Targeting Mitochondria for Treatment of Chemoresistant Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, H.; Park, S.; Yang, C.; Song, G.; Lim, W. Disruption of Endoplasmic Reticulum and ROS Production in Human Ovarian Cancer by Campesterol. Antioxidants 2021, 10, 379. https://doi.org/10.3390/antiox10030379
Bae H, Park S, Yang C, Song G, Lim W. Disruption of Endoplasmic Reticulum and ROS Production in Human Ovarian Cancer by Campesterol. Antioxidants. 2021; 10(3):379. https://doi.org/10.3390/antiox10030379
Chicago/Turabian StyleBae, Hyocheol, Sunwoo Park, Changwon Yang, Gwonhwa Song, and Whasun Lim. 2021. "Disruption of Endoplasmic Reticulum and ROS Production in Human Ovarian Cancer by Campesterol" Antioxidants 10, no. 3: 379. https://doi.org/10.3390/antiox10030379
APA StyleBae, H., Park, S., Yang, C., Song, G., & Lim, W. (2021). Disruption of Endoplasmic Reticulum and ROS Production in Human Ovarian Cancer by Campesterol. Antioxidants, 10(3), 379. https://doi.org/10.3390/antiox10030379