
Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review



Abstract
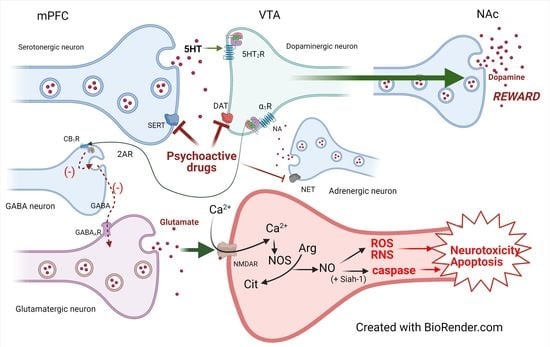
:
1. Introduction
- cocaine-3,4-methylenedioxymethamphetamine (MDMA)—mixed group, non-selective monoamine reuptake inhibitors (cocaine-like), with preferential selectivity for dopamine transporter (DAT), but favoring the release of 5-HT like MDMA (e.g., mephedrone, methylone, ethylone, naphyrone);
- methamphetamine-like cathinones group, catecholamine reuptake inhibitors and DA liberators (e.g., methcathinone, flephedrone);
- pyrovalerone-derived compounds, catecholamine reuptake inhibitors, without a liberating action (e.g., 3,4-methylenedioxypyrovalerone also known as MDPV, 3,4-methylenedioxy-α-pyrrolidinobutiophene also known as MDPBP, α-pyrrolidinovalerophenone also known as α-PVP);
2. Materials and Methods
3. Results
3.1. Transendothelial Blood Brain Barrier and Lipophilicity
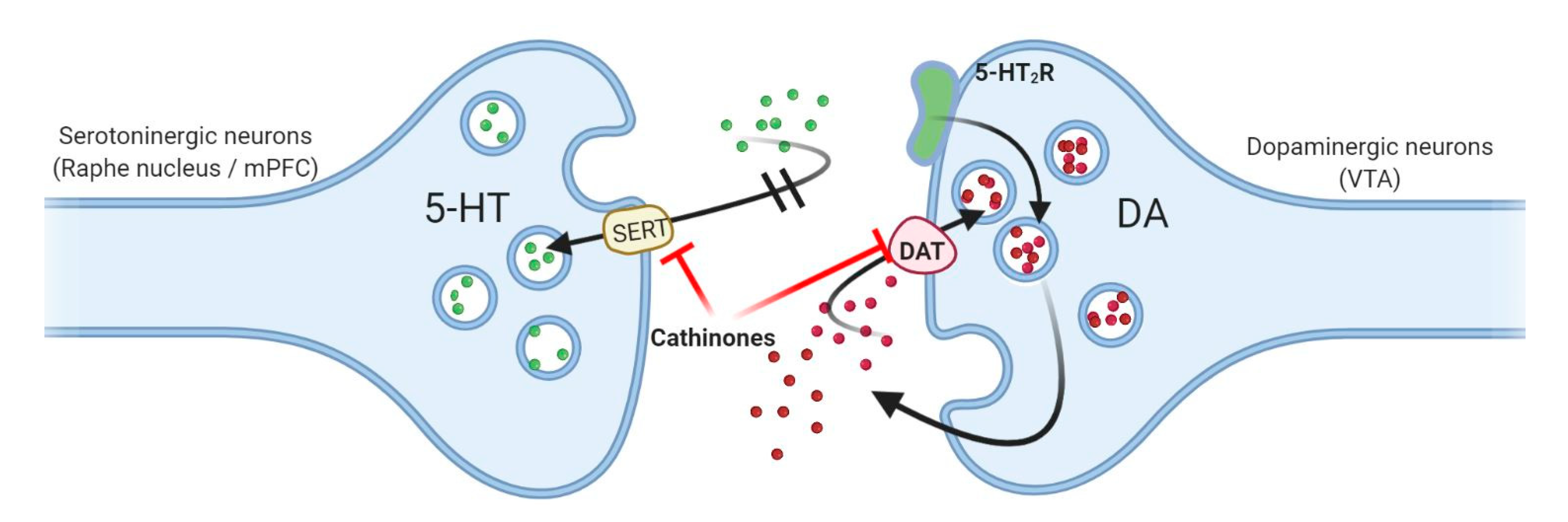
3.2. Influence of Neurotransmitter Transporters, Vesicular Transporters, and Receptors
3.3. Mechanisms of Action
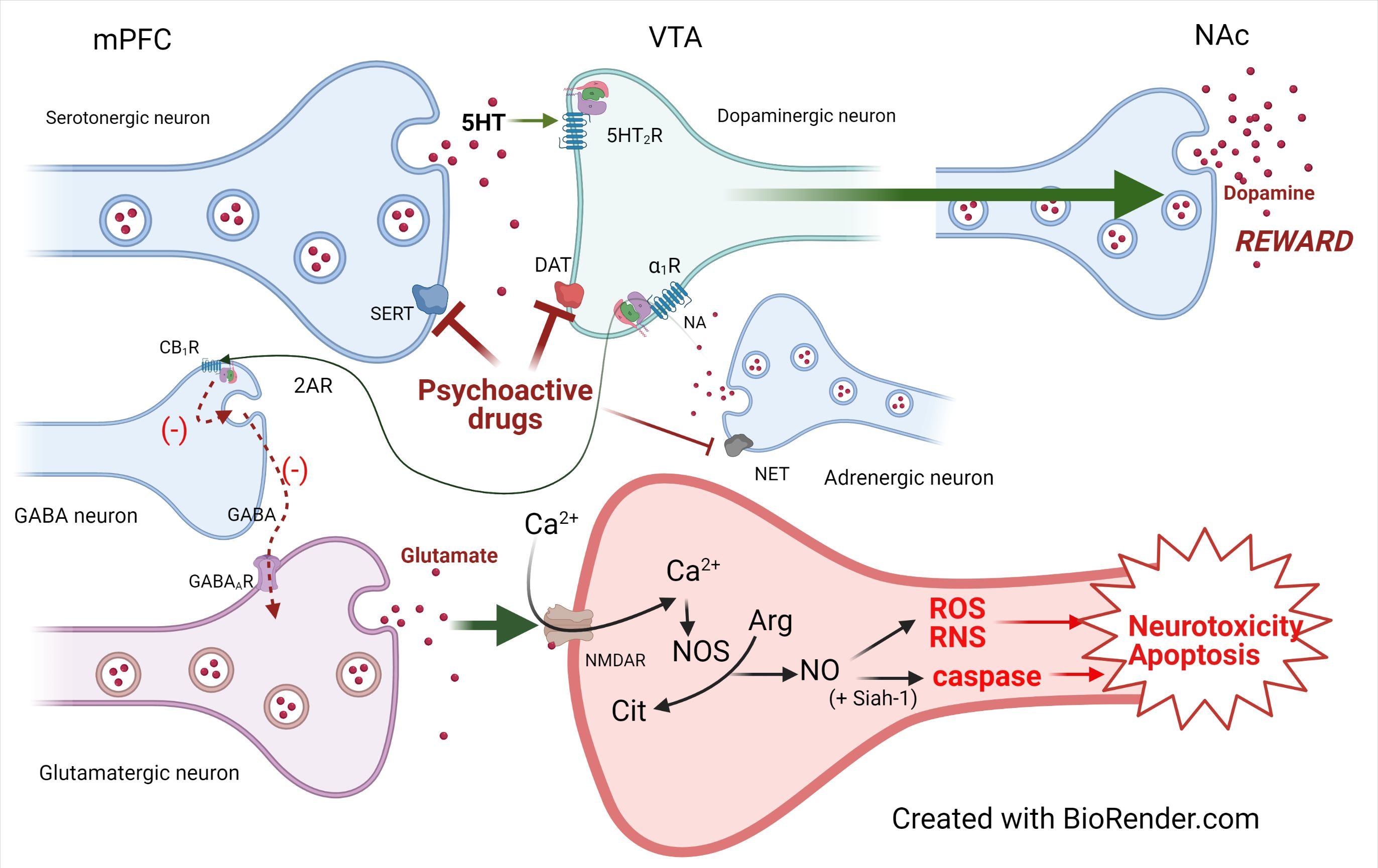
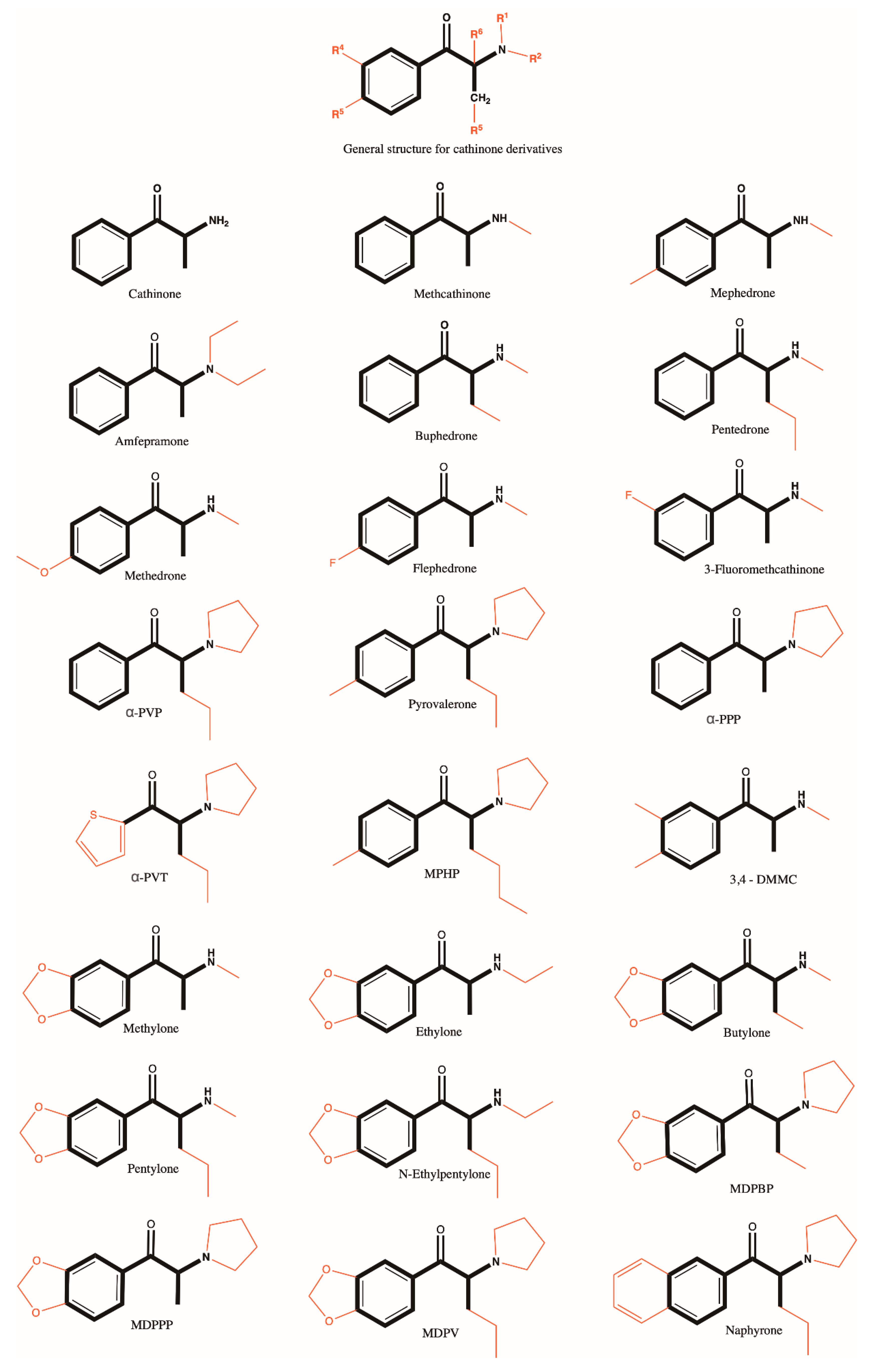
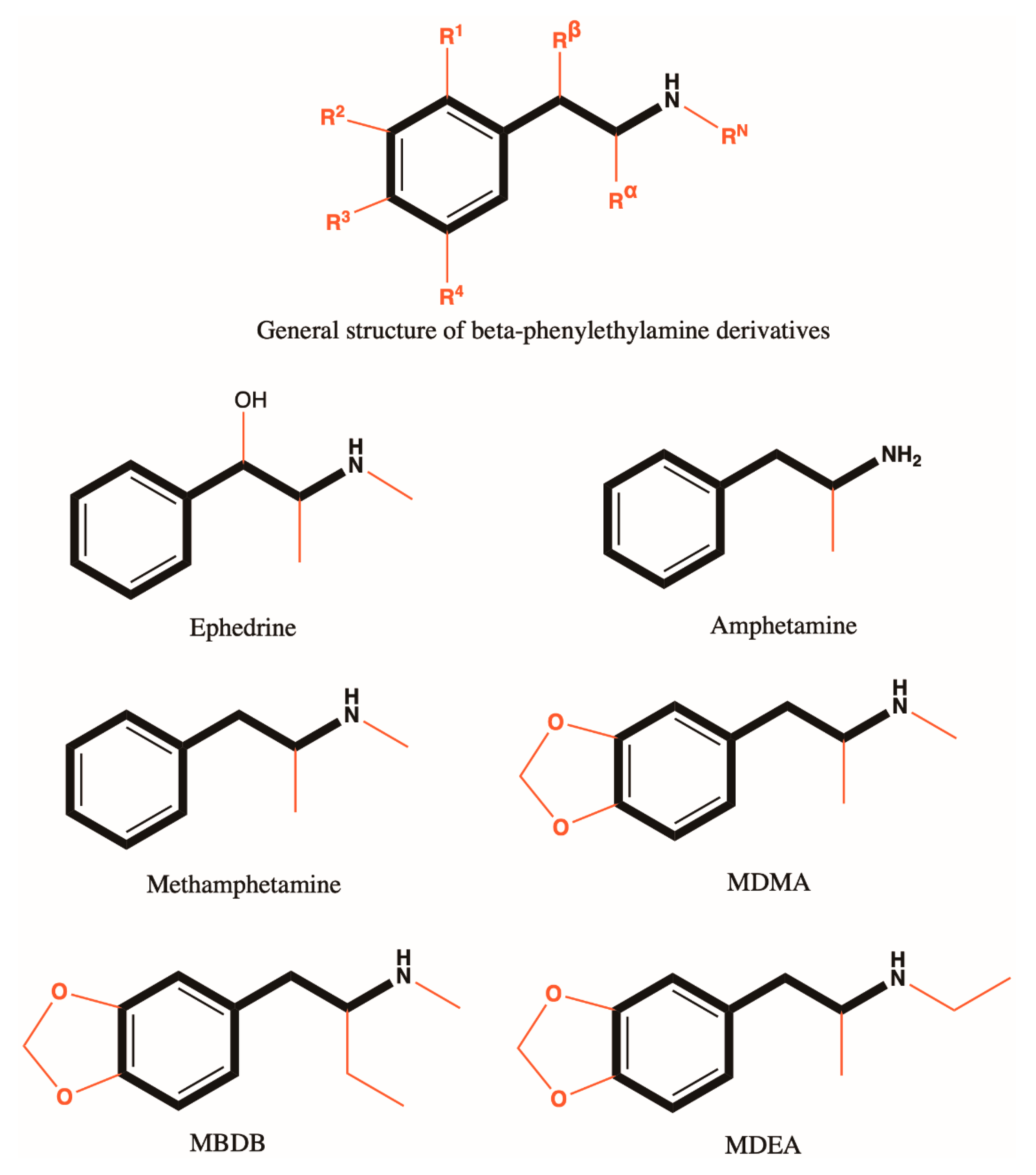
3.4. Structure Activity Relationship
3.4.1. Cathinone Derivatives
3.4.2. Amphetamine Derivatives
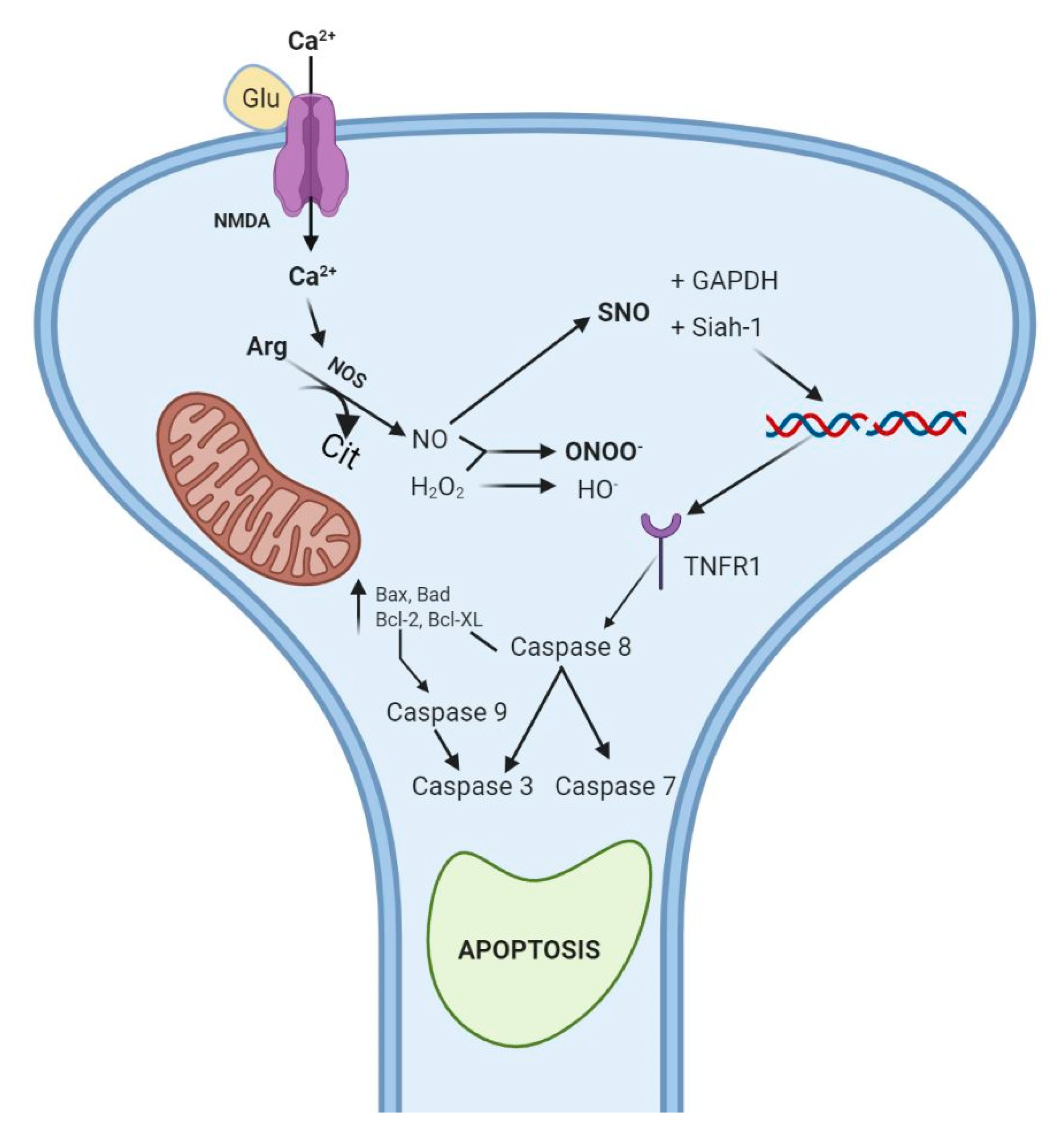
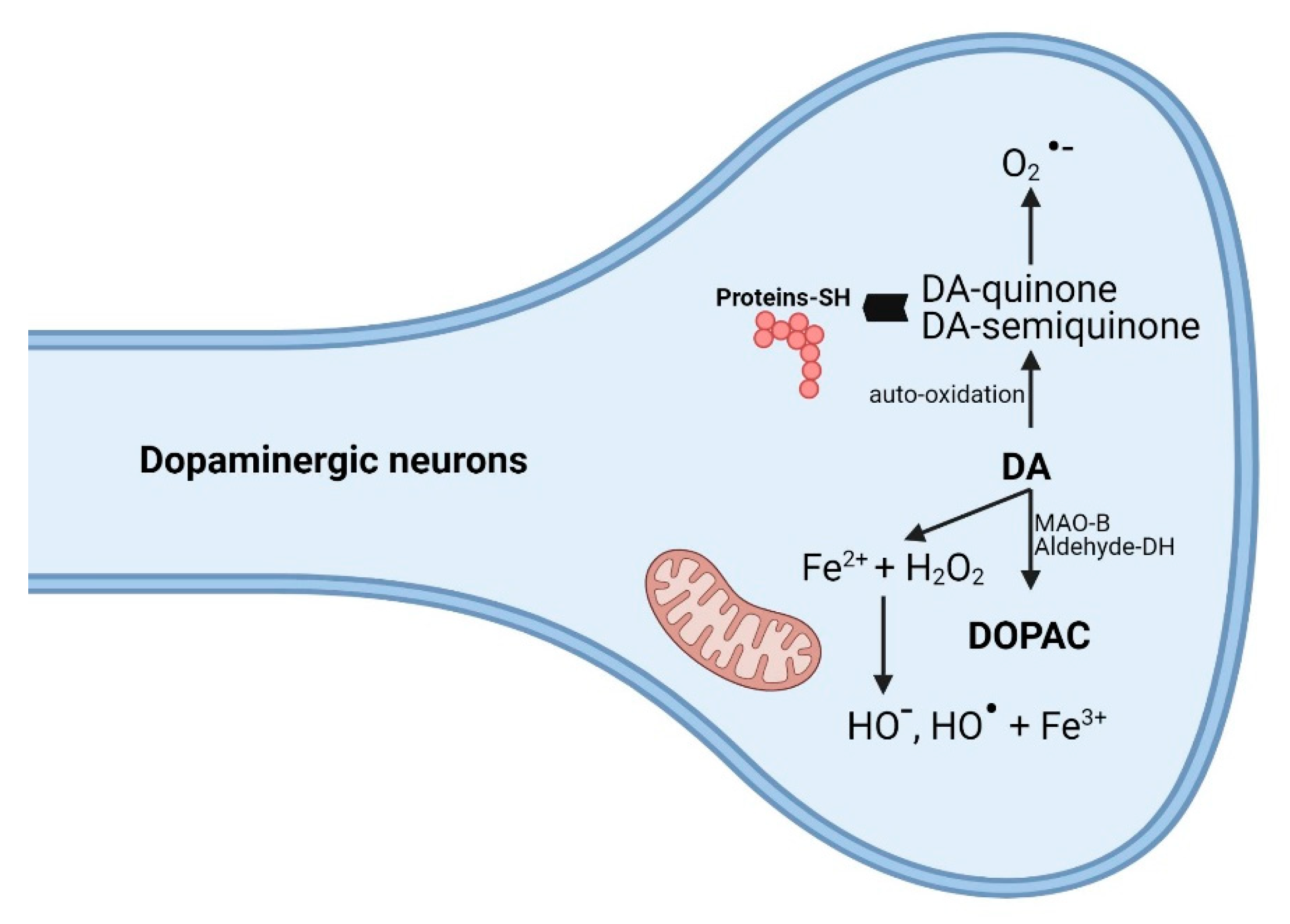
3.5. Biochemical Mechanisms: Dopamine, Oxidative Stress, and Cytotoxicity
3.6. Cathinone Derivatives
3.7. Amphetamine Derivatives
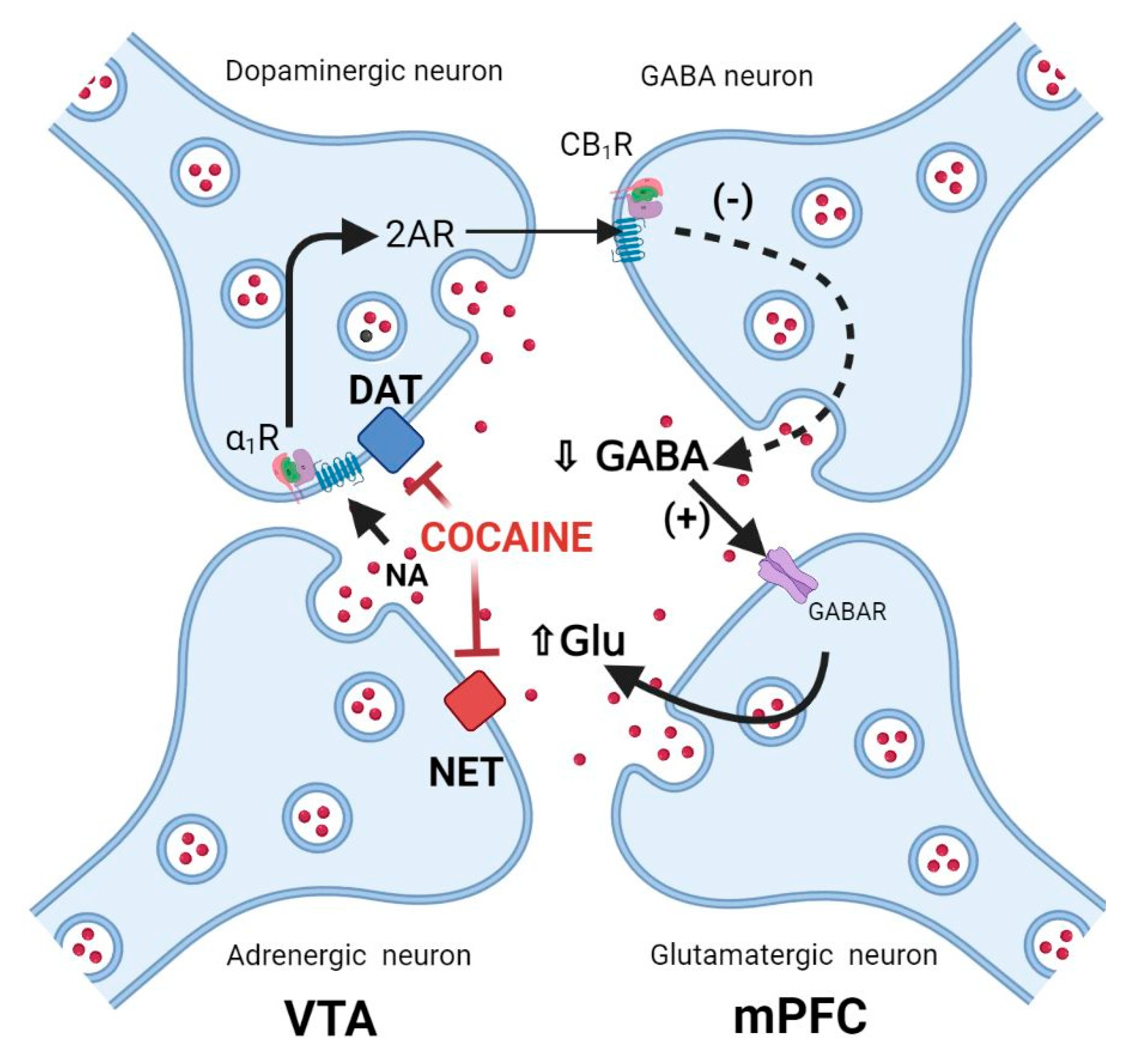
3.8. Cocaine
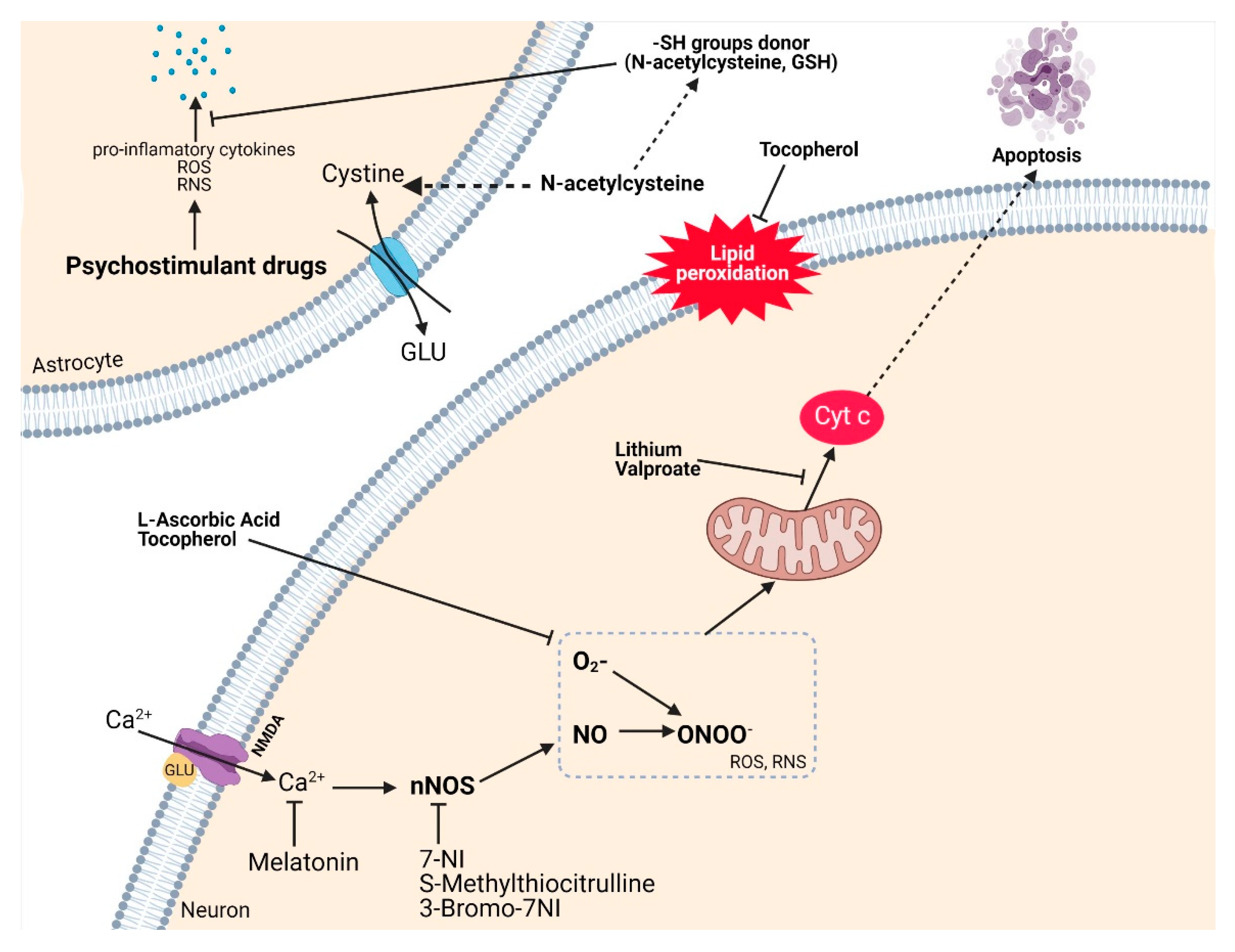
3.9. Antioxidant Therapy Related to Drug Abuse
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berman, S.; O’Neill, J.; Fears, S.; Bartzokis, G.; London, E.D. Abuse of Amphetamines and Structural Abnormalities in the Brain. Ann. N. Y. Acad. Sci. 2008, 1141, 195–220. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, B.K.; Moszczynska, A.; Gudelsky, G.A. Amphetamine Toxicities: Classical and Emerging Mechanisms. Ann. N. Y. Acad. Sci. 2010, 1187, 101–121. [Google Scholar] [CrossRef]
- Carvalho, M.; Carmo, H.; Costa, V.M.; Capela, J.P.; Pontes, H.; Remião, F.; Carvalho, F.; de Lourdes Bastos, M. Toxicity of Amphetamines: An Update. Arch. Toxicol. 2012, 86, 1167–1231. [Google Scholar] [CrossRef]
- Valente, M.J.; Guedes de Pinho, P.; de Lourdes Bastos, M.; Carvalho, F.; Carvalho, M. Khat and Synthetic Cathinones: A Review. Arch. Toxicol. 2014, 88, 15–45. [Google Scholar] [CrossRef]
- Prosser, J.M.; Nelson, L.S. The Toxicology of Bath Salts: A Review of Synthetic Cathinones. J. Med. Toxicol. 2012, 8, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreck, B.; Guerlais, M.; Laforgue, E.; Bichon, C.; Grall-Bronnec, M.; Victorri-Vigneau, C. Cathinone Use Disorder in the Context of Slam Practice: New Pharmacological and Clinical Challenges. Front. Psychiatry 2020, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Clemente, J.; Escubedo, E.; Pubill, D.; Camarasa, J. Interaction of Mephedrone with Dopamine and Serotonin Targets in Rats. Eur. Neuropsychopharmacol. 2012, 22, 231–236. [Google Scholar] [CrossRef]
- Baumann, M.H.; Ayestas, M.A.; Partilla, J.S.; Sink, J.R.; Shulgin, A.T.; Daley, P.F.; Brandt, S.D.; Rothman, R.B.; Ruoho, A.E.; Cozzi, N.V. The Designer Methcathinone Analogs, Mephedrone and Methylone, Are Substrates for Monoamine Transporters in Brain Tissue. Neuropsychopharmacology 2012, 37, 1192–1203. [Google Scholar] [CrossRef]
- Simmler, L.; Buser, T.; Donzelli, M.; Schramm, Y.; Dieu, L.-H.; Huwyler, J.; Chaboz, S.; Hoener, M.; Liechti, M. Pharmacological Characterization of Designer Cathinones in Vitro: Pharmacology of Cathinones. Br. J. Pharmacol. 2013, 168, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.H.; Partilla, J.S.; Lehner, K.R.; Thorndike, E.B.; Hoffman, A.F.; Holy, M.; Rothman, R.B.; Goldberg, S.R.; Lupica, C.R.; Sitte, H.H.; et al. Powerful Cocaine-Like Actions of 3,4-Methylenedioxypyrovalerone (MDPV), a Principal Constituent of Psychoactive ‘Bath Salts’ Products. Neuropsychopharmacology 2013, 38, 552–562. [Google Scholar] [CrossRef] [Green Version]
- López-Arnau, R.; Martínez-Clemente, J.; Pubill, D.; Escubedo, E.; Camarasa, J. Comparative Neuropharmacology of Three Psychostimulant Cathinone Derivatives: Butylone, Mephedrone and Methylone: Neuropharmacology of Cathinone Derivatives. Br. J. Pharmacol. 2012, 167, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Majchrzak, M.; Celiński, R.; Kuś, P.; Kowalska, T.; Sajewicz, M. The Newest Cathinone Derivatives as Designer Drugs: An Analytical and Toxicological Review. Forensic Toxicol. 2018, 36, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs of Abuse, A DEA Resource Guide. 2020. Available online: https://youth.gov/federal-links/resource-drugs-abuse-dea-resource-guide (accessed on 22 January 2021).
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report: Trends and Developments; Publications Office of the European Union: Luxemburg, 2020. [Google Scholar] [CrossRef]
- German, C.L.; Fleckenstein, A.E.; Hanson, G.R. Bath Salts and Synthetic Cathinones: An Emerging Designer Drug Phenomenon. Life Sci. 2014, 97, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.A.; Callahan, P.M. Monoamine Reuptake Inhibitors Enhance the Discriminative State Induced by Cocaine in the Rat. Psychopharmacology 1991, 104, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Angoa-Pérez, M.; Anneken, J.H.; Kuhn, D.M. Neurotoxicology of Synthetic Cathinone Analogs. In Neuropharmacology of New Psychoactive Substances (NPS); Baumann, M.H., Glennon, R.A., Wiley, J.L., Eds.; Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2016; Volume 32, pp. 209–230. [Google Scholar] [CrossRef]
- Kousik, S.M.; Napier, T.C.; Carvey, P.M. The Effects of Psychostimulant Drugs on Blood Brain Barrier Function and Neuroinflammation. Front. Pharmacol. 2012, 3, 121. [Google Scholar] [CrossRef] [Green Version]
- Steinkellner, T.; Montgomery, T.R.; Hofmaier, T.; Kudlacek, O.; Yang, J.-W.; Rickhag, M.; Jung, G.; Lubec, G.; Gether, U.; Freissmuth, M.; et al. Amphetamine Action at the Cocaine- and Antidepressant-Sensitive Serotonin Transporter Is Modulated by CaMKII. J. Neurosci. 2015, 35, 8258–8271. [Google Scholar] [CrossRef] [Green Version]
- Fantegrossi, W.E.; Ciullo, J.R.; Wakabayashi, K.T.; De La Garza, R.; Traynor, J.R.; Woods, J.H. A Comparison of the Physiological, Behavioral, Neurochemical and Microglial Effects of Methamphetamine and 3,4-Methylenedioxymethamphetamine in the Mouse. Neuroscience 2008, 151, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Krasnova, I.N.; Justinova, Z.; Ladenheim, B.; Jayanthi, S.; McCoy, M.T.; Barnes, C.; Warner, J.E.; Goldberg, S.R.; Cadet, J.L. Methamphetamine Self-Administration Is Associated with Persistent Biochemical Alterations in Striatal and Cortical Dopaminergic Terminals in the Rat. PLoS ONE 2010, 5, e8790. [Google Scholar] [CrossRef] [Green Version]
- Shaerzadeh, F.; Streit, W.J.; Heysieattalab, S.; Khoshbouei, H. Methamphetamine Neurotoxicity, Microglia, and Neuroinflammation. J. Neuroinflamm. 2018, 15, 341. [Google Scholar] [CrossRef]
- Glennon, R.A.; Yousif, M.; Naiman, N.; Kalix, P. Methcathinone: A New and Potent Amphetamine-like Agent. Pharmacol. Biochem. Behav. 1987, 26, 547–551. [Google Scholar] [CrossRef]
- Glennon, R.A.; Dukat, M. Structure-Activity Relationships of Synthetic Cathinones. In Neuropharmacology of New Psychoactive Substances (NPS); Baumann, M.H., Glennon, R.A., Wiley, J.L., Eds.; Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2016; Volume 32, pp. 19–47. [Google Scholar] [CrossRef]
- Kehr, J.; Ichinose, F.; Yoshitake, S.; Goiny, M.; Sievertsson, T.; Nyberg, F.; Yoshitake, T. Mephedrone, Compared with MDMA (Ecstasy) and Amphetamine, Rapidly Increases Both Dopamine and 5-HT Levels in Nucleus Accumbens of Awake Rats: Mephedrone Increases Both DA and 5-HT in Rat Brain. Br. J. Pharmacol. 2011, 164, 1949–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, M.J.; Angrish, D.; Aarde, S.M.; Barlow, D.J.; Buczynski, M.W.; Creehan, K.M.; Vandewater, S.A.; Parsons, L.H.; Houseknecht, K.L.; Dickerson, T.J.; et al. Effect of Ambient Temperature on the Thermoregulatory and Locomotor Stimulant Effects of 4-Methylmethcathinone in Wistar and Sprague-Dawley Rats. PLoS ONE 2012, 7, e44652. [Google Scholar] [CrossRef] [PubMed]
- Shortall, S.E.; Spicer, C.H.; Ebling, F.J.P.; Green, A.R.; Fone, K.C.F.; King, M.V. Contribution of Serotonin and Dopamine to Changes in Core Body Temperature and Locomotor Activity in Rats Following Repeated Administration of Mephedrone: Repeated Mephedrone Effects. Addict. Biol. 2016, 21, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Salam, O.M.E.; Morsy, S.M.Y.; Youness, E.R.; Yassen, N.N.; Sleem, A.A. The Effect of Low Dose Amphetamine in Rotenone-Induced Toxicity in a Mice Model of Parkinson’s Disease. Iran. J. Basic Med. Sci. 2020, 23, 1207. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Solis, E.; Watterson, L.R.; Marusich, J.A.; Fantegrossi, W.E.; Wiley, J.L. Baths Salts, Spice, and Related Designer Drugs: The Science Behind the Headlines. J. Neurosci. 2014, 34, 15150–15158. [Google Scholar] [CrossRef] [Green Version]
- Cameron, K.; Kolanos, R.; Verkariya, R.; De Felice, L.; Glennon, R.A. Mephedrone and Methylenedioxypyrovalerone (MDPV), Major Constituents of “Bath Salts,” Produce Opposite Effects at the Human Dopamine Transporter. Psychopharmacology 2013, 227, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Naoi, M.; Maruyama, W. Cell Death of Dopamine Neurons in Aging and Parkinson’s Disease. Mech. Ageing Dev. 1999, 111, 175–188. [Google Scholar] [CrossRef]
- Soto-Otero, R.; Munoz-Patino, A.M.; Labandeira-Garcia, J.L. Autoxidation and Neurotoxicity of 6-Hydroxydopamine in the Presence of Some Antioxidants: Potential Implication in Relation to the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2000, 74, 8. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Approaches to Prevent Dopamine Quinone-Induced Neurotoxicity. Neurochem. Res. 2009, 34, 698–706. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Gu, H.H.; Zhan, C.-G. Mechanism for Cocaine Blocking the Transport of Dopamine: Insights from Molecular Modeling and Dynamics Simulations. J. Phys. Chem. B 2009, 113, 15057–15066. [Google Scholar] [CrossRef] [Green Version]
- Cocaine and Amphetamine-like Psychostimulants: Neurocircuitry and Glutamate Neuroplasticity. Dialogues Clin. Neurosci. 2007, 9, 389–397. [CrossRef]
- Robertson, S.D.; Matthies, H.J.G.; Galli, A. A Closer Look at Amphetamine-Induced Reverse Transport and Trafficking of the Dopamine and Norepinephrine Transporters. Mol. Neurobiol. 2009, 39, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Daberkow, D.P.; Brown, H.D.; Bunner, K.D.; Kraniotis, S.A.; Doellman, M.A.; Ragozzino, M.E.; Garris, P.A.; Roitman, M.F. Amphetamine Paradoxically Augments Exocytotic Dopamine Release and Phasic Dopamine Signals. J. Neurosci. 2013, 33, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.H.; Ayestas, M.A.; Sharpe, L.G.; Lewis, D.B.; Rice, K.C.; Rothman, R.B. Persistent Antagonism of Methamphetamine-Induced Dopamine Release in Rats Pretreated with GBR12909 Decanoate. J. Pharmacol. Exp. Ther. 2002, 301, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Partilla, J.S.; Dempsey, A.G.; Nagpal, A.S.; Blough, B.E.; Baumann, M.H.; Rothman, R.B. Interaction of Amphetamines and Related Compounds at the Vesicular Monoamine Transporter. J. Pharmacol. Exp. Ther. 2006, 319, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, K.R.; Block, E.R.; Levitan, E.S. Action Potentials and Amphetamine Release Antipsychotic Drug from Dopamine Neuron Synaptic VMAT Vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E4485–E4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyberg, Z.; Sonders, M.S.; Aguilar, J.I.; Hiranita, T.; Karam, C.S.; Flores, J.; Pizzo, A.B.; Zhang, Y.; Farino, Z.J.; Chen, A.; et al. Mechanisms of Amphetamine Action Illuminated through Optical Monitoring of Dopamine Synaptic Vesicles in Drosophila Brain. Nat. Commun. 2016, 7, 10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huotari, M.; García-Horsman, J.A.; Karayiorgou, M.; Gogos, J.A.; Männistö, P.T. D-Amphetamine Responses in Catechol-O-Methyltransferase (COMT) Disrupted Mice. Psychopharmacology 2004, 172, 1–10. [Google Scholar] [CrossRef]
- Liu, C.H.; Ren, J.; Liu, P.K. Amphetamine Manipulates Monoamine Oxidase-A Level and Behavior Using Theranostic Aptamers of Transcription Factors AP-1/NF-KB. J. Biomed. Sci. 2016, 23, 21. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Parada, M.; Iturriaga-Vasquez, P.; Cassels, B.K. Amphetamine Derivatives as Monoamine Oxidase Inhibitors. Front. Pharmacol. 2020, 10, 1590. [Google Scholar] [CrossRef]
- Juárez Olguín, H.; Calderón Guzmán, D.; Hernández García, E.; Barragán Mejía, G. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxid. Med. Cell. Longev. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anneken, J.H.; Angoa-Pérez, M.; Kuhn, D.M. 3,4-Methylenedioxypyrovalerone Prevents While Methylone Enhances Methamphetamine-Induced Damage to Dopamine Nerve Endings: β-Ketoamphetamine Modulation of Neurotoxicity by the Dopamine Transporter. J. Neurochem. 2015, 133, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Clemente, J.; López-Arnau, R.; Carbó, M.; Pubill, D.; Camarasa, J.; Escubedo, E. Mephedrone Pharmacokinetics after Intravenous and Oral Administration in Rats: Relation to Pharmacodynamics. Psychopharmacology 2013, 229, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.-S.; Reith, M.E.A.; Yan, S. Enhanced Accumbal Dopamine Release Following 5-HT2A Receptor Stimulation in Rats Pretreated with Intermittent Cocaine. Brain Res. 2000, 863, 254–258. [Google Scholar] [CrossRef]
- Auclair, A.; Drouin, C.; Cotecchia, S.; Glowinski, J.; Tassin, J.-P. 5-HT2A and A1b-Adrenergic Receptors Entirely Mediate Dopamine Release, Locomotor Response and Behavioural Sensitization to Opiates and Psychostimulants: 5-HT2A and A1b-AR Mediate Behavioural Sensitization. Eur. J. Neurosci. 2004, 20, 3073–3084. [Google Scholar] [CrossRef]
- Del Bello, F.; Sakloth, F.; Partilla, J.S.; Baumann, M.H.; Glennon, R.A. Ethylenedioxy Homologs of N-Methyl-(3,4-Methylenedioxyphenyl)-2-Aminopropane (MDMA) and Its Corresponding Cathinone Analog Methylenedioxymethcathinone: Interactions with Transporters for Serotonin, Dopamine, and Norepinephrine. Bioorg. Med. Chem. 2015, 23, 5574–5579. [Google Scholar] [CrossRef] [Green Version]
- Marusich, J.A.; Antonazzo, K.R.; Wiley, J.L.; Blough, B.E.; Partilla, J.S.; Baumann, M.H. Pharmacology of Novel Synthetic Stimulants Structurally Related to the “Bath Salts” Constituent 3,4-Methylenedioxypyrovalerone (MDPV). Neuropharmacology 2014, 87, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, P.C.; Butler, D.; Deschamps, J.R.; Madras, B.K. 1-(4-Methylphenyl)-2-Pyrrolidin-1-Yl-Pentan-1-One (Pyrovalerone) Analogues: A Promising Class of Monoamine Uptake Inhibitors. J. Med. Chem. 2006, 49, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshleman, A.J.; Wolfrum, K.M.; Hatfield, M.G.; Johnson, R.A.; Murphy, K.V.; Janowsky, A. Substituted Methcathinones Differ in Transporter and Receptor Interactions. Biochem. Pharmacol. 2013, 85, 1803–1815. [Google Scholar] [CrossRef] [Green Version]
- Eshleman, A.J.; Wolfrum, K.M.; Reed, J.F.; Kim, S.O.; Swanson, T.; Johnson, R.A.; Janowsky, A. Structure-Activity Relationships of Substituted Cathinones, with Transporter Binding, Uptake, and Release. J. Pharmacol. Exp. Ther. 2017, 360, 33–47. [Google Scholar] [CrossRef]
- Baumann, M.H.; Walters, H.M.; Niello, M.; Sitte, H.H. Neuropharmacology of Synthetic Cathinones. In New Psychoactive Substances; Handbook of Experimental Pharmacology; Maurer, H.H., Brandt, S.D., Eds.; Springer: Cham, Switzerland, 2018; Volume 252, pp. 113–142. [Google Scholar] [CrossRef]
- Tyrkkö, E.; Pelander, A.; Ketola, R.A.; Ojanperä, I. In Silico and in Vitro Metabolism Studies Support Identification of Designer Drugs in Human Urine by Liquid Chromatography/Quadrupole-Time-of-Flight Mass Spectrometry. Anal. Bioanal. Chem. 2013, 405, 6697–6709. [Google Scholar] [CrossRef] [PubMed]
- Sakloth, F.; Kolanos, R.; Mosier, P.D.; Bonano, J.S.; Banks, M.L.; Partilla, J.S.; Baumann, M.H.; Negus, S.S.; Glennon, R.A. Steric Parameters, Molecular Modeling and Hydropathic Interaction Analysis of the Pharmacology of Para-Substituted Methcathinone Analogues: Further QSAR of Para-Substituted Methcathinone Analogues. Br. J. Pharmacol. 2015, 172, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swortwood, M.J.; Carlier, J.; Ellefsen, K.N.; Wohlfarth, A.; Diao, X.; Concheiro-Guisan, M.; Kronstrand, R.; Huestis, M.A. In Vitro, in Vivo and in Silico Metabolic Profiling of α-Pyrrolidinopentiothiophenone, a Novel Thiophene Stimulant. Bioanalysis 2016, 8, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Ellefsen, K.N.; Wohlfarth, A.; Swortwood, M.J.; Diao, X.; Concheiro, M.; Huestis, M.A. 4-Methoxy-α-PVP: In Silico Prediction, Metabolic Stability, and Metabolite Identification by Human Hepatocyte Incubation and High-Resolution Mass Spectrometry. Forensic Toxicol. 2016, 34, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niello, M.; Cintulova, D.; Hellsberg, E.; Jäntsch, K.; Holy, M.; Ayatollahi, L.H.; Cozzi, N.V.; Freissmuth, M.; Sandtner, W.; Ecker, G.F.; et al. Para-Trifluoromethyl-Methcathinone Is an Allosteric Modulator of the Serotonin Transporter. Neuropharmacology 2019, 161, 107615. [Google Scholar] [CrossRef]
- Duart-Castells, L.; Nadal-Gratacós, N.; Muralter, M.; Puster, B.; Berzosa, X.; Estrada-Tejedor, R.; Niello, M.; Bhat, S.; Pubill, D.; Camarasa, J.; et al. Role of Amino Terminal Substitutions in the Pharmacological, Rewarding and Psychostimulant Profiles of Novel Synthetic Cathinones. Neuropharmacology 2021, 186, 108475. [Google Scholar] [CrossRef]
- Negus, S.S.; Banks, M.L. Decoding the Structure of Abuse Potential for New Psychoactive Substances: Structure–Activity Relationships for Abuse-Related Effects of 4-Substituted Methcathinone Analogs. In Neuropharmacology of New Psychoactive Substances (NPS); Baumann, M.H., Glennon, R.A., Wiley, J.L., Eds.; Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2016; Volume 32, pp. 119–131. [Google Scholar] [CrossRef]
- Rickli, A.; Hoener, M.C.; Liechti, M.E. Monoamine Transporter and Receptor Interaction Profiles of Novel Psychoactive Substances: Para-Halogenated Amphetamines and Pyrovalerone Cathinones. Eur. Neuropsychopharmacol. 2015, 25, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Zwartsen, A.; Olijhoek, M.E.; Westerink, R.H.S.; Hondebrink, L. Hazard Characterization of Synthetic Cathinones Using Viability, Monoamine Reuptake, and Neuronal Activity Assays. Front. Neurosci. 2020, 14, 9. [Google Scholar] [CrossRef]
- Luethi, D.; Kolaczynska, K.E.; Docci, L.; Krähenbühl, S.; Hoener, M.C.; Liechti, M.E. Pharmacological Profile of Mephedrone Analogs and Related New Psychoactive Substances. Neuropharmacology 2018, 134, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Simmler, L.D.; Rickli, A.; Hoener, M.C.; Liechti, M.E. Monoamine Transporter and Receptor Interaction Profiles of a New Series of Designer Cathinones. Neuropharmacology 2014, 79, 152–160. [Google Scholar] [CrossRef]
- Saha, K.; Li, Y.; Holy, M.; Lehner, K.R.; Bukhari, M.O.; Partilla, J.S.; Sandtner, W.; Sitte, H.H.; Baumann, M.H. The Synthetic Cathinones, Butylone and Pentylone, Are Stimulants That Act as Dopamine Transporter Blockers but 5-HT Transporter Substrates. Psychopharmacology 2019, 236, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Partilla, J.S.; Lehner, K.R.; Seddik, A.; Stockner, T.; Holy, M.; Sandtner, W.; Ecker, G.F.; Sitte, H.H.; Baumann, M.H. ‘Second-Generation’ Mephedrone Analogs, 4-MEC and 4-MePPP, Differentially Affect Monoamine Transporter Function. Neuropsychopharmacology 2015, 40, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Banks, M.L.; Worst, T.J.; Rusyniak, D.E.; Sprague, J.E. Synthetic Cathinones (“Bath Salts”). J. Emerg. Med. 2014, 46, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J. 3,4-Methylenedioxymethamphetamine (MDMA): Current Perspectives. Subst. Abuse Rehabil. 2013, 4, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, M.G.; Lee, R.; Wardle, M.C.; Jacob, S.; de Wit, H. Effects of MDMA and Intranasal Oxytocin on Social and Emotional Processing. Neuropsychopharmacology 2014, 39, 1654–1663. [Google Scholar] [CrossRef] [Green Version]
- Rothman, R.B.; Vu, N.; Partilla, J.S.; Roth, B.L.; Hufeisen, S.J.; Compton-Toth, B.A.; Birkes, J.; Young, R.; Glennon, R.A. In Vitro Characterization of Ephedrine-Related Stereoisomers at Biogenic Amine Transporters and the Receptorome Reveals Selective Actions as Norepinephrine Transporter Substrates. J. Pharmacol. Exp. Ther. 2003, 307, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Cozzi, N.V.; Brandt, S.D.; Daley, P.F.; Partilla, J.S.; Rothman, R.B.; Tulzer, A.; Sitte, H.H.; Baumann, M.H. Pharmacological Examination of Trifluoromethyl Ring-Substituted Methcathinone Analogs. Eur. J. Pharmacol. 2013, 699, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Puerta, E.; Aguirre, N. Methylenedioxymethamphetamine (MDMA, ’Ecstasy’): Neurodegeneration versus Neuromodulation. Pharmaceuticals 2011, 4, 992–1018. [Google Scholar] [CrossRef] [Green Version]
- Haber, S.N.; Heilbronner, S.R. Translational Research in OCD: Circuitry and Mechanisms. Neuropsychopharmacology 2013, 38, 252. [Google Scholar] [CrossRef] [Green Version]
- Parrott, A.C. Recreational Ecstasy/MDMA, the Serotonin Syndrome, and Serotonergic Neurotoxicity. Pharmacol. Biochem. Behav. 2002, 71, 837–844. [Google Scholar] [CrossRef]
- de Bie, R.M.A.; Gladstone, R.M.; Strafella, A.P.; Ko, J.-H.; Lang, A.E. Manganese-Induced Parkinsonism Associated With Methcathinone (Ephedrone) Abuse. Arch. Neurol. 2007, 64, 886. [Google Scholar] [CrossRef] [Green Version]
- Stepens, A.; Logina, I.; Liguts, V.; Aldiņš, P.; Ekšteina, I.; Platkājis, A.; Mārtiņsone, I.; Tērauds, E.; Rozentāle, B.; Donaghy, M. A Parkinsonian Syndrome in Methcathinone Users and the Role of Manganese. N. Engl. J. Med. 2008, 358, 1009–1017. [Google Scholar] [CrossRef]
- Selikhova, M.; Fedoryshyn, L.; Matviyenko, Y.; Komnatska, I.; Kyrylchuk, M.; Krolicki, L.; Friedman, A.; Taylor, A.; Jäger, H.R.; Lees, A.; et al. Parkinsonism and Dystonia Caused by the Illicit Use of Ephedrone—A Longitudinal Study. Mov. Disord. 2008, 23, 2224–2231. [Google Scholar] [CrossRef]
- Fudalej, S.; Kołodziejczyk, I.; Gajda, T.; Majkowska-Zwolińska, B.; Wojnar, M. Manganese-Induced Parkinsonism among Ephedrone Users and Drug Policy in Poland. J. Addict. Med. 2013, 7, 302–303. [Google Scholar] [CrossRef]
- Debruyne, D.; Loilier, M.; Cesbron, A.; Le Boisselier, R.; Bourgine, J. Emerging Drugs of Abuse: Current Perspectives on Substituted Cathinones. Subst. Abuse Rehabil. 2014, 5, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Schifano, F.; Napoletano, F.; Arillotta, D.; Zangani, C.; Gilgar, L.; Guirguis, A.; Corkery, J.M.; Vento, A. The Clinical Challenges of Synthetic Cathinones. Br. J. Clin. Pharmacol. 2020, 86, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Tero-Vescan, A.; Vari, C.-E.; Imre, S.; Filip, C.; Hancu, G. Comparative Analysis by HPLC-UV and Capillary Electrophoresis of Dietary Supplements for Weight Loss. Farmacia 2016, 64, 699–705. [Google Scholar]
- Collins, G.T.; Abbott, M.; Galindo, K.; Rush, E.L.; Rice, K.C.; France, C.P. Discriminative Stimulus Effects of Binary Drug Mixtures: Studies with Cocaine, MDPV, and Caffeine. J. Pharmacol. Exp. Ther. 2016, 359, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirguis, A.; Corkery, J.M.; Stair, J.L.; Kirton, S.B.; Zloh, M.; Schifano, F. Intended and Unintended Use of Cathinone Mixtures. Hum. Psychopharmacol. Clin. Exp. 2017, 32, e2598. [Google Scholar] [CrossRef]
- Gannon, B.M.; Galindo, K.I.; Mesmin, M.P.; Rice, K.C.; Collins, G.T. Reinforcing Effects of Binary Mixtures of Common Bath Salt Constituents: Studies with 3,4-Methylenedioxypyrovalerone (MDPV), 3,4-Methylenedioxymethcathinone (Methylone), and Caffeine in Rats. Neuropsychopharmacology 2018, 43, 761–769. [Google Scholar] [CrossRef]
- Schiavone, S.; Jaquet, V.; Trabace, L.; Krause, K.-H. Severe Life Stress and Oxidative Stress in the Brain: From Animal Models to Human Pathology. Antioxid. Redox Signal. 2013, 18, 1475–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Dee, C.M.; Shen, J. Interaction of Free Radicals, Matrix Metalloproteinases and Caveolin-1 Impacts Blood-Brain Barrier Permeability. Front. Biosci. 2011, 3, 1216–1231. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative Stress and Apoptosis. Pathophisiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Kanti Das, T.; Wati, M.R.; Fatima-Shad, K. Oxidative Stress Gated by Fenton and Haber Weiss Reactions and Its Association with Alzheimer’s Disease. Arch. Neurosci. 2014, 2, e60038. [Google Scholar] [CrossRef] [Green Version]
- Pomierny-Chamioło, L.; Moniczewski, A.; Wydra, K.; Suder, A.; Filip, M. Oxidative Stress Biomarkers in Some Rat Brain Structures and Peripheral Organs Underwent Cocaine. Neurotox. Res. 2013, 23, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorce, S.; Krause, K.-H. NOX Enzymes in the Central Nervous System: From Signaling to Disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C. Pleasure Systems in the Brain. Neuron 2015, 86, 646–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, P.; Dinan, T.G.; Psychopharmacol, J. Prolactin and Dopamine: What Is the Connection? A Review. J. Psyschopharmacol. 2008, 22, 12–19. [Google Scholar] [CrossRef]
- Marchetti, B.; L’Episcopo, F.; Morale, M.C.; Tirolo, C.; Testa, N.; Caniglia, S.; Serapide, M.F.; Pluchino, S. Uncovering Novel Actors in Astrocyte-Neuron Crosstalk in Parkinson’s Disease: The Wnt/β-Catenin Signaling Cascade as the Common Final Pathway for Neuroprotection and Self-Repair. Eur. J. Neurosci. 2013, 37, 1550–1563. [Google Scholar] [CrossRef] [Green Version]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Paul, R.; Choudhury, A.; Kumar, S.; Giri, A.; Sandhir, R.; Borah, A. Cholesterol Contributes to Dopamine-Neuronal Loss in MPTP Mouse Model of Parkinson’s Disease: Involvement of Mitochondrial Dysfunctions and Oxidative Stress. PLoS ONE 2017, 12, e0171285. [Google Scholar] [CrossRef] [Green Version]
- Angoa-Pérez, M.; Kane, M.J.; Briggs, D.I.; Francescutti, D.M.; Sykes, C.E.; Shah, M.M.; Thomas, D.M.; Kuhn, D.M. Mephedrone Does Not Damage Dopamine Nerve Endings of the Striatum, but Enhances the Neurotoxicity of Methamphetamine, Amphetamine, and MDMA. J. Neurochem. 2013, 125, 102–110. [Google Scholar] [CrossRef]
- Angoa-Pérez, M.; Kane, M.J.; Francescutti, D.M.; Sykes, K.E.; Shah, M.M.; Mohammed, A.M.; Thomas, D.M.; Kuhn, D.M. Mephedrone, an Abused Psychoactive Component of ‘Bath Salts’ and Methamphetamine Congener, Does Not Cause Neurotoxicity to Dopamine Nerve Endings of the Striatum: Mephedrone Does Not Cause DA Nerve Terminal Toxicity. J. Neurochem. 2012, 120, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Suyama, J.A.; Sakloth, F.; Kolanos, R.; Glennon, R.A.; Lazenka, M.F.; Negus, S.S.; Banks, M.L. Abuse-Related Neurochemical Effects of Para-Substituted Methcathinone Analogs in Rats: Microdialysis Studies of Nucleus Accumbens Dopamine and Serotonin. J. Pharmacol. Exp. Ther. 2015, 356, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Hadlock, G.C.; Webb, K.M.; McFadden, L.M.; Chu, P.W.; Ellis, J.D.; Allen, S.C.; Andrenyak, D.M.; Vieira-Brock, P.L.; German, C.L.; Conrad, K.M.; et al. 4-Methylmethcathinone (Mephedrone): Neuropharmacological Effects of a Designer Stimulant of Abuse. J. Pharmacol. Exp. Ther. 2011, 339, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Motbey, C.P.; Karanges, E.; Li, K.M.; Wilkinson, S.; Winstock, A.R.; Ramsay, J.; Hicks, C.; Kendig, M.D.; Wyatt, N.; Callaghan, P.D.; et al. Mephedrone in Adolescent Rats: Residual Memory Impairment and Acute but Not Lasting 5-HT Depletion. PLoS ONE 2012, 7, e45473. [Google Scholar] [CrossRef] [Green Version]
- den Hollander, B.; Rozov, S.; Linden, A.-M.; Uusi-Oukari, M.; Ojanperä, I.; Korpi, E.R. Long-Term Cognitive and Neurochemical Effects of “Bath Salt” Designer Drugs Methylone and Mephedrone. Pharmacol. Biochem. Behav. 2013, 103, 501–509. [Google Scholar] [CrossRef]
- López-Arnau, R.; Martínez-Clemente, J.; Abad, S.; Pubill, D.; Camarasa, J.; Escubedo, E. Repeated Doses of Methylone, a New Drug of Abuse, Induce Changes in Serotonin and Dopamine Systems in the Mouse. Psychopharmacology 2014, 231, 3119–3129. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Clemente, J.; López-Arnau, R.; Abad, S.; Pubill, D.; Escubedo, E.; Camarasa, J. Dose and Time-Dependent Selective Neurotoxicity Induced by Mephedrone in Mice. PLoS ONE 2014, 9, e99002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Arnau, R.; Martínez-Clemente, J.; Rodrigo, T.; Pubill, D.; Camarasa, J.; Escubedo, E. Neuronal Changes and Oxidative Stress in Adolescent Rats after Repeated Exposure to Mephedrone. Toxicol. Appl. Pharmacol. 2015, 286, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Pantano, F.; Tittarelli, R.; Mannocchi, G.; Pacifici, R.; di Luca, A.; Busardò, F.P.; Marinelli, E. Neurotoxicity Induced by Mephedrone: An up-to-Date Review. Curr. Neuropharmacol. 2017, 15, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamińska, K.; Noworyta-Sokołowska, K.; Górska, A.; Rzemieniec, J.; Wnuk, A.; Wojtas, A.; Kreiner, G.; Kajta, M.; Gołembiowska, K. The Effects of Exposure to Mephedrone During Adolescence on Brain Neurotransmission and Neurotoxicity in Adult Rats. Neurotox. Res. 2018, 34, 525–537. [Google Scholar] [CrossRef]
- Escubedo, E.; Chipana, C.; Pérez-Sánchez, M.; Camarasa, J.; Pubill, D. Methyllycaconitine Prevents Methamphetamine-Induced Effects in Mouse Striatum: Involvement of A7 Nicotinic Receptors. J. Pharmacol. Exp. Ther. 2005, 315, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Chipana, C.; García-Ratés, S.; Camarasa, J.; Pubill, D.; Escubedo, E. Different Oxidative Profile and Nicotinic Receptor Interaction of Amphetamine and 3,4-Methylenedioxy-Methamphetamine. Neurochem. Int. 2008, 52, 401–410. [Google Scholar] [CrossRef]
- Potula, R.; Hawkins, B.J.; Cenna, J.M.; Fan, S.; Dykstra, H.; Ramirez, S.H.; Morsey, B.; Brodie, M.R.; Persidsky, Y. Methamphetamine Causes Mitrochondrial Oxidative Damage in Human T Lymphocytes Leading to Functional Impairment. J. Immunol. 2010, 185, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Leong, H.S.; Philp, M.; Simone, M.; Witting, P.K.; Fu, S. Synthetic Cathinones Induce Cell Death in Dopaminergic SH-SY5Y Cells via Stimulating Mitochondrial Dysfunction. Int. J. Mol. Sci. 2020, 21, 1370. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.-J.; Tran, H.-Q.; Nguyen, P.-T.; Jeong, J.H.; Nah, S.-Y.; Jang, C.-G.; Nabeshima, T.; Kim, H.-C. Role of Mitochondria in Methamphetamine-Induced Dopaminergic Neurotoxicity: Involvement in Oxidative Stress, Neuroinflammation, and Pro-Apoptosis—A Review. Neurochem. Res. 2018, 43, 66–78. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 437–454. [Google Scholar] [CrossRef]
- Marvanova, M. Drug-Induced Cognitive Impairment: Effect of Cardiovascular Agents. Ment. Health Clin. 2016, 6, 201–206. [Google Scholar] [CrossRef]
- Budzynska, B.; Boguszewska-Czubara, A.; Kruk-Slomka, M.; Kurzepa, J.; Biala, G. Mephedrone and Nicotine: Oxidative Stress and Behavioral Interactions in Animal Models. Neurochem. Res. 2015, 40, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Safhi, M.M.; Alam, M.F.; Hussain, S.; Hakeem Siddiqui, M.A.; Khuwaja, G.; Jubran Khardali, I.A.; Al-Sanosi, R.M.; Islam, F. Cathinone, an Active Principle of Catha Edulis, Accelerates Oxidative Stress in the Limbic Area of Swiss Albino Mice. J. Ethnopharmacol. 2014, 156, 102–106. [Google Scholar] [CrossRef]
- Fitzmaurice, P.S.; Tong, J.; Yazdanpanah, M.; Liu, P.P.; Kalasinsky, K.S.; Kish, S.J. Levels of 4-Hydroxynonenal and Malondialdehyde Are Increased in Brain of Human Chronic Users of Methamphetamine. J. Pharmacol. Exp. Ther. 2006, 319, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Sikk, K.; Taba, P.; Haldre, S.; Bergquist, J.; Nyholm, D.; Zjablov, G.; Asser, T.; Aquilonius, S.-M. Irreversible Motor Impairment in Young Addicts? Ephedrone, Manganism or Both? Acta Neurol. Scand. 2007, 115, 385–389. [Google Scholar] [CrossRef]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxid. Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormoehlen, L.M.; Kumar, N. Neurotoxicology: Five New Things. Neurol. Clin. Pract. 2012, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.G.; Cooney, P.T.; Erikson, K.M. Inhibition of DAT Function Attenuates Manganese Accumulation in the Globus Pallidus. Environ. Toxicol. Pharmacol. 2007, 23, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Verina, T.; Kiihl, S.F.; Schneider, J.S.; Guilarte, T.R. Manganese Exposure Induces Microglia Activation and Dystrophy in the Substantia Nigra of Non-Human Primates. NeuroToxicology 2011, 32, 215–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepens, A.; Stagg, C.J.; Platkājis, A.; Boudrias, M.-H.; Johansen-Berg, H.; Donaghy, M. White Matter Abnormalities in Methcathinone Abusers with an Extrapyramidal Syndrome. Brain 2010, 133, 3676–3684. [Google Scholar] [CrossRef] [PubMed]
- Stepens, A.; Groma, V.; Skuja, S.; Platkājis, A.; Aldiņš, P.; Ekšteina, I.; Mārtiņsone, I.; Bricis, R.; Donaghy, M. The Outcome of the Movement Disorder in Methcathinone Abusers: Clinical, MRI and Manganesemia Changes, and Neuropathology. Eur. J. Neurol. 2014, 21, 199–205. [Google Scholar] [CrossRef]
- Fasano, A. Methcathinone-Induced Parkinsonism Results from Permanent Brain Damage: A Message for the Masses. Eur. J. Neurol. 2014, 21, 181–182. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Burton, N.C.; McGlothan, J.L.; Verina, T.; Zhou, Y.; Alexander, M.; Pham, L.; Griswold, M.; Wong, D.F.; Syversen, T.; et al. Impairment of Nigrostriatal Dopamine Neurotransmission by Manganese Is Mediated by Pre-Synaptic Mechanism(s): Implications to Manganese-Induced Parkinsonism. J. Neurochem. 2008, 107, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anneken, J.H.; Angoa-Perez, M.; Sati, G.C.; Crich, D.; Kuhn, D.M. Assessing the Role of Dopamine in the Differential Neurotoxicity Patterns of Methamphetamine, Mephedrone, Methcathinone and 4-Methylmethamphetamine. Neuropharmacology 2018, 134, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.N.; Martins, M.R.; Petronilho, F.C.; Dal-Pizzol, F.; Quevedo, J.; Kapczinski, F. Increased Oxidative Stress after Repeated Amphetamine Exposure: Possible Relevance as a Model of Mania. Bipolar Disord. 2006, 8, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Granado, N.; Ares-Santos, S.; Oliva, I.; O’Shea, E.; Martin, E.D.; Colado, M.I.; Moratalla, R. Dopamine D2-Receptor Knockout Mice Are Protected against Dopaminergic Neurotoxicity Induced by Methamphetamine or MDMA. Neurobiol. Dis. 2011, 42, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Mirecki, A.; Fitzmaurice, P.; Ang, L.; Kalasinsky, K.S.; Peretti, F.J.; Aiken, S.S.; Wickham, D.J.; Sherwin, A.; Nobrega, J.N.; Forman, H.J.; et al. Brain Antioxidant Systems in Human Methamphetamine Users: Brain Oxidative Stress in Methamphetamine Users. J. Neurochem. 2004, 89, 1396–1408. [Google Scholar] [CrossRef]
- Liang, H.-W.; Xia, Q.; Bruce, I.C. Reactive Oxygen Species Mediate the Neuroprotection Conferred by a Mitochondrial ATP-Sensitive Potassium Channel Opener during Ischemia in the Rat Hippocampal Slice. Brain Res. 2005, 1042, 169–175. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A Molecular Switch Node in the Crosstalk between Autophagy and Apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Cadet, J.L.; Jayanthi, S.; Deng, X. Methamphetamine-Induced Neuronal Apoptosis Involves the Activation of Multiple Death Pathways. Review. Neurotox. Res. 2005, 8, 199–206. [Google Scholar] [CrossRef]
- Gonçalves, J.; Baptista, S.; Silva, A.P. Psychostimulants and Brain Dysfunction: A Review of the Relevant Neurotoxic Effects. Neuropharmacology 2014, 87, 135–149. [Google Scholar] [CrossRef]
- Brown, J.M.; Yamamoto, B.K. Effects of Amphetamines on Mitochondrial Function: Role of Free Radicals and Oxidative Stress. Pharmacol. Ther. 2003, 99, 45–53. [Google Scholar] [CrossRef]
- Suto, N.; Ecke, L.E.; You, Z.-B.; Wise, R.A. Extracellular Fluctuations of Dopamine and Glutamate in the Nucleus Accumbens Core and Shell Associated with Lever-Pressing during Cocaine Self-Administration, Extinction, and Yoked Cocaine Administration. Psychopharmacology 2010, 211, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, E.E.; Brock, M.V.; Lange, M.S.; Troncoso, J.C.; Blue, M.E.; Lowenstein, C.J.; Johnston, M.V.; Baumgartner, W.A. Glutamate Excitotoxicity Mediates Neuronal Apoptosis After Hypothermic Circulatory Arrest. Ann. Thorac. Surg. 2010, 89, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Michaelis, E.K. Selective Neuronal Vulnerability to Oxidative Stress in the Brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Treadway, T.; Covey, D.P.; Cheer, J.F.; Lupica, C.R. Cocaine-Induced Endocannabinoid Mobilization in the Ventral Tegmental Area. Cell Rep. 2015, 12, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.L.; Krasnova, I.N.; Jayanthi, S.; Lyles, J. Neurotoxicity of Substituted Amphetamines: Molecular and Cellular Mechanisms. Neurotox. Res. 2007, 11, 183–202. [Google Scholar] [CrossRef]
- Pubill, D.; Chipana, C.; Camins, A.; Pallàs, M.; Camarasa, J.; Escubedo, E. Free Radical Production Induced by Methamphetamine in Rat Striatal Synaptosomes. Toxicol. Appl. Pharmacol. 2005, 204, 57–68. [Google Scholar] [CrossRef]
- Frey, B.N.; Valvassori, S.S.; Gomes, K.M.; Martins, M.R.; Dal-Pizzol, F.; Kapczinski, F.; Quevedo, J. Increased Oxidative Stress in Submitochondrial Particles after Chronic Amphetamine Exposure. Brain Res. 2006, 1097, 224–229. [Google Scholar] [CrossRef]
- Bashkatova, V.; Mathieu-Kia, A.-M.; Durand, C.; Penit-Soria, J. Neurochemical Changes and Neurotoxic Effects of an Acute Treatment with Sydnocarb, A Novel Psychostimulant: Comparison with d-Amphetamine. Ann. N. Y. Acad. Sci. 2006, 965, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Serritella, A.V.; Sen, T.; Farook, J.M.; Sedlak, T.W.; Baraban, J.; Snyder, S.H.; Sen, N. Behavioral Effects of Cocaine Mediated by Nitric Oxide-GAPDH Transcriptional Signaling. Neuron 2013, 78, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Chen, L.; Liu, C.; Qiu, P.; Wang, A.; Li, L.; Wang, H. Up-Regulation of Protein Tyrosine Nitration in Methamphetamine-Induced Neurotoxicity through DDAH/ADMA/NOS Pathway. Neurochem. Int. 2013, 62, 1055–1064. [Google Scholar] [CrossRef]
- Tsang, A.H.K.; Chung, K.K.K. Oxidative and Nitrosative Stress in Parkinson’s Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2009, 1792, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Cui, K.; Luo, X.; Xu, K.; Ven Murthy, M.R. Role of Oxidative Stress in Neurodegeneration: Recent Developments in Assay Methods for Oxidative Stress and Nutraceutical Antioxidants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 771–799. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Raudensky, J. The Role of Oxidative Stress, Metabolic Compromise, and Inflammation in Neuronal Injury Produced by Amphetamine-Related Drugs of Abuse. J. Neuroimmune Pharmacol. 2008, 3, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Berman, S.B.; Hastings, T.G. Dopamine Oxidation Alters Mitochondrial Respiration and Induces Permeability Transition in Brain Mitochondria: Implications for Parkinson’s Disease. J. Neurochem. 2001, 73, 1127–1137. [Google Scholar] [CrossRef]
- Pereira, R.B.; Andrade, P.B.; Valentão, P. A Comprehensive View of the Neurotoxicity Mechanisms of Cocaine and Ethanol. Neurotox. Res. 2015, 28, 253–267. [Google Scholar] [CrossRef]
- Muriach, M.; López-Pedrajas, R.; Barcia, J.M.; Sanchez-Villarejo, M.V.; Almansa, I.; Romero, F.J. Cocaine Causes Memory and Learning Impairments in Rats: Involvement of Nuclear Factor Kappa B and Oxidative Stress, and Prevention by Topiramate: Topiramate Effects on Cocaine Administration. J. Neurochem. 2010, 114, 675–684. [Google Scholar] [CrossRef]
- Kovacic, P. Role of Oxidative Metabolites of Cocaine in Toxicity and Addiction: Oxidative Stress and Electron Transfer. Med. Hypotheses 2005, 64, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Öztezcan, S.; Doğru-Abbasoğlu, S.; Mutlu-Türkoğlu, Ü.; Calay, Z.; Aykaç-Toker, G.; Uysal, M. The Role of Stimulated Lipid Peroxidation and Impaired Calcium Sequestration in the Enhancement of Cocaine Induced Hepatotoxicity by Ethanol. Drug Alcohol Depend. 2000, 58, 77–83. [Google Scholar] [CrossRef]
- Poon, H.F.; Abdullah, L.; Mullan, M.A.; Mullan, M.J.; Crawford, F.C. Cocaine-Induced Oxidative Stress Precedes Cell Death in Human Neuronal Progenitor Cells. Neurochem. Int. 2007, 50, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Uys, J.D. Cocaine-Induced Adaptations in Cellular Redox Balance Contributes to Enduring Behavioral Plasticity. Neuropsychopharmacology 2011, 36, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Ghosh, J.; Sil, P. Drug Metabolism and Oxidative Stress: Cellular Mechanism and New Therapeutic Insights. Biochem. Anal. Biochem. 2016, 5, 255. [Google Scholar] [CrossRef]
- López-Pedrajas, R.; Ramírez-Lamelas, D.T.; Muriach, B.; Sánchez-Villarejo, M.V.; Almansa, I.; Vidal-Gil, L.; Romero, F.J.; Barcia, J.M.; Muriach, M. Cocaine Promotes Oxidative Stress and Microglial-Macrophage Activation in Rat Cerebellum. Front. Cell. Neurosci. 2015, 9, 279. [Google Scholar] [CrossRef] [Green Version]
- Beiser, T.; Yaka, R. The Role of Oxidative Stress in Cocaine Addiction. J. Neurol. Neuromed. 2019, 4, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Macêdo, D.S.; de Vasconcelos, S.M.M.; dos Santos, R.S.; Aguiar, L.M.V.; Lima, V.T.M.; Viana, G.S.B.; de Sousa, F.C.F. Cocaine Alters Catalase Activity in Prefrontal Cortex and Striatum of Mice. Neurosci. Lett. 2005, 387, 53–56. [Google Scholar] [CrossRef]
- Jang, E.Y.; Yang, C.H.; Hedges, D.M.; Kim, S.P.; Lee, J.Y.; Ekins, T.G.; Garcia, B.T.; Kim, H.Y.; Nelson, A.C.; Kim, N.J.; et al. The Role of Reactive Oxygen Species in Methamphetamine Self-Administration and Dopamine Release in the Nucleus Accumbens: ROS in METH-Taking Behavior. Addict. Biol. 2017, 22, 1304–1315. [Google Scholar] [CrossRef]
- Walker, J.; Winhusen, T.; Storkson, J.M.; Lewis, D.; Pariza, M.W.; Somoza, E.; Somoza, V. Total Antioxidant Capacity Is Significantly Lower in Cocaine-Dependent and Methamphetamine-Dependent Patients Relative to Normal Controls: Results from a Preliminary Study: Oxidative stress and stimulant dependence. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Little, K.Y.; Ramssen, E.; Welchko, R.; Volberg, V.; Roland, C.J.; Cassin, B. Decreased Brain Dopamine Cell Numbers in Human Cocaine Users. Psychiatry Res. 2009, 168, 173–180. [Google Scholar] [CrossRef]
- Sordi, A.O.; Pechansky, F.; Kessler, F.H.P.; Kapczinski, F.; Pfaffenseller, B.; Gubert, C.; de Aguiar, B.W.; de Magalhães Narvaez, J.C.; Ornell, F.; von Diemen, L. Oxidative Stress and BDNF as Possible Markers for the Severity of Crack Cocaine Use in Early Withdrawal. Psychopharmacology 2014, 231, 4031–4039. [Google Scholar] [CrossRef]
- Berríos-Cárcamo, P.; Quezada, M.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y.; Ezquer, F. Oxidative Stress and Neuroinflammation as a Pivot in Drug Abuse. A Focus on the Therapeutic Potential of Antioxidant and Anti-Inflammatory Agents and Biomolecules. Antioxidants 2020, 9, 830. [Google Scholar] [CrossRef] [PubMed]
- Thrash-Williams, B.; Karuppagounder, S.S.; Bhattacharya, D.; Ahuja, M.; Suppiramaniam, V.; Dhanasekaran, M. Methamphetamine-Induced Dopaminergic Toxicity Prevented Owing to the Neuroprotective Effects of Salicylic Acid. Life Sci. 2016, 154, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-K.T.; Lee, J.; Shin, E.-J.; Dang, D.-K.; Jeong, J.H.; Nguyen, T.-T.L.; Nam, Y.; Cho, H.-J.; Lee, J.-C.; Park, D.H.; et al. Liposomal Melatonin Rescues Methamphetamine-Elicited Mitochondrial Burdens, pro-Apoptosis, and Dopaminergic Degeneration through the Inhibition PKCδ Gene. J. Pineal Res. 2015, 58, 86–106. [Google Scholar] [CrossRef]
- Huang, Y.-N.; Wang, J.-Y.; Lee, C.-T.; Lin, C.-H.; Lai, C.-C.; Wang, J.-Y. L-Ascorbate Attenuates Methamphetamine Neurotoxicity through Enhancing the Induction of Endogenous Heme Oxygenase-1. Toxicol. Appl. Pharmacol. 2012, 265, 241–252. [Google Scholar] [CrossRef]
- Huang, Y.-N.; Yang, L.-Y.; Wang, J.-Y.; Lai, C.-C.; Chiu, C.-T.; Wang, J.-Y. L-Ascorbate Protects Against Methamphetamine-Induced Neurotoxicity of Cortical Cells via Inhibiting Oxidative Stress, Autophagy, and Apoptosis. Mol. Neurobiol. 2017, 54, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Perera, J.; Tan, J.; Jeevathayaparan, S.; Chakravarthi, S.; Haleagrahara, N. Neuroprotective Effects of Alpha Lipoic Acid on Haloperidol-Induced Oxidative Stress in the Rat Brain. Cell Biosci. 2011, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moszczynska, A.; Yamamoto, B.K. Methamphetamine Oxidatively Damages Parkin and Decreases the Activity of 26S Proteasome in Vivo: Methamphetamine and Ubiquitin-Proteasome System. J. Neurochem. 2011, 116, 1005–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, S.Z.; Newport, G.D.; Islam, F.; Slikker, W.; Ali, S.F. Selenium, an Antioxidant, Protects against Methamphetamine-Induced Dopaminergic Neurotoxicity. Brain Res. 1999, 818, 575–578. [Google Scholar] [CrossRef]
- Barayuga, S.M.; Pang, X.; Andres, M.A.; Panee, J.; Bellinger, F.P. Methamphetamine Decreases Levels of Glutathione Peroxidases 1 and 4 in SH-SY5Y Neuronal Cells: Protective Effects of Selenium. NeuroToxicology 2013, 37, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Ungvári, É.; Monori, I.; Megyeri, A.; Csiki, Z.; Prokisch, J.; Sztrik, A.; Jávor, A.; Benkő, I. Protective Effects of Meat from Lambs on Selenium Nanoparticle Supplemented Diet in a Mouse Model of Polycyclic Aromatic Hydrocarbon-Induced Immunotoxicity. Food Chem. Toxicol. 2014, 64, 298–306. [Google Scholar] [CrossRef]
- Ghosh, P.; Bhattacharjee, A.; Basu, A.; Singha Roy, S.; Bhattacharya, S. Attenuation of Cyclophosphamide-Induced Pulmonary Toxicity in Swiss Albino Mice by Naphthalimide-Based Organoselenium Compound 2-(5-Selenocyanatopentyl)-Benzo[de]Isoquinoline 1,3-Dione. Pharm. Biol. 2015, 53, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Kiełczykowska, M.; Kocot, J.; Paździor, M.; Musik, I. Selenium—A Fascinating Antioxidant of Protective Properties. Adv. Clin. Exp. Med. 2018, 27, 245–255. [Google Scholar] [CrossRef]
- Bachmann, R.F.; Wang, Y.; Yuan, P.; Zhou, R.; Li, X.; Alesci, S.; Du, J.; Manji, H.K. Common Effects of Lithium and Valproate on Mitochondrial Functions: Protection against Methamphetamine-Induced Mitochondrial Damage. Int. J. Neuropsychopharmacol. 2009, 12, 805. [Google Scholar] [CrossRef] [Green Version]
- Feier, G.; Valvassori, S.S.; Varela, R.B.; Resende, W.R.; Bavaresco, D.V.; Morais, M.O.; Scaini, G.; Andersen, M.L.; Streck, E.L.; Quevedo, J. Lithium and Valproate Modulate Energy Metabolism in an Animal Model of Mania Induced by Methamphetamine. Pharmacol. Biochem. Behav. 2013, 103, 589–596. [Google Scholar] [CrossRef]
- Imam, S.Z.; Islam, F.; Itzhak, Y.; Slikker, W.; Ali, S.F. Prevention of Dopaminergic Neurotoxicity by Targeting Nitric Oxide and Peroxynitrite: Implications for the Prevention of Methamphetamine-Induced Neurotoxic Damage. Ann. N. Y. Acad. Sci. 2000, 914, 157–171. [Google Scholar] [CrossRef]
- Di Monte, D.A.; Royland, J.E.; Jakowec, M.W.; Langston, J.W. Role of Nitric Oxide in Methamphetamine Neurotoxicity: Protection by 7-Nitroindazole, an Inhibitor of Neuronal Nitric Oxide Synthase. J. Neurochem. 2002, 67, 2443–2450. [Google Scholar] [CrossRef]
- Virmani, A.; Gaetani, F.; Imam, S.; Binienda, Z.; Ali, S. Possible Mechanism for the Neuroprotective Effects of L-Carnitine on Methamphetamine-Evoked Neurotoxicity. Ann. N. Y. Acad. Sci. 2003, 993, 197–207. [Google Scholar] [CrossRef]
- Itzhak, Y.; Martin, J.L.; Ali, S.F. NNOS Inhibitors Attenuate Methamphetamine-Induced Dopaminergic Neurotoxicity but Not Hyperthermia in Mice. NeuroReport 2000, 11, 2943–2946. [Google Scholar] [CrossRef]
- Suwanjang, W.; Abramov, A.Y.; Charngkaew, K.; Govitrapong, P.; Chetsawang, B. Melatonin Prevents Cytosolic Calcium Overload, Mitochondrial Damage and Cell Death Due to Toxically High Doses of Dexamethasone-Induced Oxidative Stress in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Int. 2016, 97, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Pi, H.; Zhang, L.; Zhang, N.; Li, Y.; Zhang, H.; Tang, J.; Li, H.; Feng, M.; Deng, P.; et al. Melatonin Prevents Abnormal Mitochondrial Dynamics Resulting from the Neurotoxicity of Cadmium by Blocking Calcium-Dependent Translocation of Drp1 to the Mitochondria. J. Pineal Res. 2016, 60, 291–302. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tsukada, H.; Nishiyama, S.; Fukumoto, D.; Kakiuchi, T.; Shimizu, E.; Iyo, M. Protective Effects of N-Acetyl-L-Cysteine on the Reduction of Dopamine Transporters in the Striatum of Monkeys Treated with Methamphetamine. Neuropsychopharmacology 2004, 29, 2018–2023. [Google Scholar] [CrossRef]
- Zhang, X.; Banerjee, A.; Banks, W.A.; Ercal, N. N-Acetylcysteine Amide Protects against Methamphetamine-Induced Oxidative Stress and Neurotoxicity in Immortalized Human Brain Endothelial Cells. Brain Res. 2009, 1275, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.E.; Odlaug, B.L.; Kim, S.W. A Double-Blind, Placebo-Controlled Study of N-Acetyl Cysteine plus Naltrexone for Methamphetamine Dependence. Eur. Neuropsychopharmacol. 2010, 20, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tobwala, S.; Ercal, N. N -Acetylcysteine Amide Protects against Methamphetamine-Induced Tissue Damage in CD-1 Mice. Hum. Exp. Toxicol. 2012, 31, 931–944. [Google Scholar] [CrossRef]
- Chandramani Shivalingappa, P.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A. N-Acetyl Cysteine Protects against Methamphetamine-Induced Dopaminergic Neurodegeneration via Modulation of Redox Status and Autophagy in Dopaminergic Cells. Parkinson’s Dis. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- McKetin, R.; Dean, O.M.; Baker, A.L.; Carter, G.; Turner, A.; Kelly, P.J.; Berk, M. A Potential Role for N-Acetylcysteine in the Management of Methamphetamine Dependence: N-Acetylcysteine for Methamphetamine. Drug Alcohol Rev. 2017, 36, 153–159. [Google Scholar] [CrossRef]
- Fukami, G.; Hashimoto, K.; Koike, K.; Okamura, N.; Shimizu, E.; Iyo, M. Effect of Antioxidant N-Acetyl-l-Cysteine on Behavioral Changes and Neurotoxicity in Rats after Administration of Methamphetamine. Brain Res. 2004, 1016, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Baptista, S.; Bento, A.R.; Gonçalves, J.; Bernardino, L.; Summavielle, T.; Lobo, A.; Fontes-Ribeiro, C.; Malva, J.O.; Agasse, F.; Silva, A.P. Neuropeptide Y Promotes Neurogenesis and Protection against Methamphetamine-Induced Toxicity in Mouse Dentate Gyrus-Derived Neurosphere Cultures. Neuropharmacology 2012, 62, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Moreno, A.Y.; Aarde, S.M.; Creehan, K.M.; Vandewater, S.A.; Vaillancourt, B.D.; Wright, M.J.; Janda, K.D.; Taffe, M.A. A Methamphetamine Vaccine Attenuates Methamphetamine-Induced Disruptions in Thermoregulation and Activity in Rats. Biol. Psychiatry 2013, 73, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambuchen, M.D.; Carroll, F.I.; Rüedi-Bettschen, D.; Hendrickson, H.P.; Hennings, L.J.; Blough, B.E.; Brieaddy, L.E.; Pidaparthi, R.R.; Owens, S.M. Combining Active Immunization with Monoclonal Antibody Therapy To Facilitate Early Initiation of a Long-Acting Anti-Methamphetamine Antibody Response. J. Med. Chem. 2015, 58, 4665–4677. [Google Scholar] [CrossRef] [Green Version]
- Baracz, S.J.; Cornish, J.L. The Neurocircuitry Involved in Oxytocin Modulation of Methamphetamine Addiction. Front. Neuroendocrinol. 2016, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Wu, K.-L.; Tsai, H.-M.; Chen, C.-H. Treatment of Methamphetamine Abuse: An Antibody-Based Immunotherapy Approach. J. Food Drug Anal. 2013, 21, S82–S86. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.L.; Hasalliu, E.; Hasalliu, M.; Angulo, J.A. Epigallocatechin Gallate Mitigates the Methamphetamine-Induced Striatal Dopamine Terminal Toxicity by Preventing Oxidative Stress in the Mouse Brain. Neurotox. Res. 2020, 37, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, J.; Zhang, J.; Fujita, Y.; Ishima, T.; Iyo, M.; Hashimoto, K. Protective Effects of the Antioxidant Sulforaphane on Behavioral Changes and Neurotoxicity in Mice after the Administration of Methamphetamine. Psychopharmacology 2012, 222, 37–45. [Google Scholar] [CrossRef]
- Wąsik, A.; Antkiewicz-Michaluk, L. The Mechanism of Neuroprotective Action of Natural Compounds. Pharmacol. Rep. 2017, 69, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kanthasamy, K.; Gordon, R.; Jin, H.; Anantharam, V.; Ali, S.; G. Kanthasamy, A.; Kanthasamy, A. Neuroprotective Effect of Resveratrol Against Methamphetamine-Induced Dopaminergic Apoptotic Cell Death in a Cell Culture Model of Neurotoxicity. Curr. Neuropharmacol. 2011, 9, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. 7,8-Dihydroxyflavone, a Small-Molecule TrkB Agonist, Reverses Memory Deficits and BACE1 Elevation in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Zhang, J.-C.; Ma, M.; Fujita, Y.; Wu, J.; Hashimoto, K. 7,8-Dihydroxyflavone, a TrkB Agonist, Attenuates Behavioral Abnormalities and Neurotoxicity in Mice after Administration of Methamphetamine. Psychopharmacology 2014, 231, 159–166. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Monoamine Uptake Transporter Inhibition (IC50) | Monoamine Release (EC50) | ||||||
|---|---|---|---|---|---|---|---|---|
| DAT | SERT | DAT/SERT Ratio | Ref. | DAT | SERT | DAT/SERT Ratio | Ref. | |
| Cathinone | 14.0 (10–20) * | >100 * | >10 * | [9] | 5.64 (3.0–10) * | >100 * | na | [9] |
| Methcathinone | 1.12 (0.83–1.5) * | >10 * | >10 * | [9] | 2.36 (1.7–3.3) * | >33 * | na | [9] |
| 2.4 (1.7–3.4) * | 46 (30–71) * | 19 (8.8–42) * | [64] | 12.5 ** | 3.860 ** | 309 ** | [56] | |
| Mephedrone | 3.31 (2.6–4.2) * | 4.64 (3.7–5.9) * | 1.4 (0.9–2.4) * | [9] | 3.75 (1.7–8.4) * | 5.98 (3.2–11) * | na | [9] |
| 1.4 (1.2–1.4) * | 83 (66–104) | na | [65] | 49.1 ± 8.32 ** | 118.3 ± 25.9 ** | 2.41 ** | [10] | |
| 5.7 (4.5–7.2) * | 3.6 (2.8–4.6) * | 0.63 (0.39–1.02) * | [66] | |||||
| 970 ± 50 ** | 310 ± 80 ** | na | [7] | |||||
| 762 ± 79 ** | 422 ± 26 ** | na | [10] | 51 ± 5 ** | 122 ± 10 ** | na | [10] | |
| Buphedrone | 4.24 (3.3–5.5) * | 70 (2–2700) * | >10 * | [67] | ||||
| Pentedrone | 2.5 (2.0–3.2) * | 135 (5–3700) * | >10 * | [67] | >100 * | >100 * | na | [55] |
| 0.4 (0.3–0.4) * | 16 (14–18) * | na | [65] | |||||
| Methedrone | 35 (15–79) * | 4.73(3.2–6.9) * | 0.14 (0.04–0.46) * | [67] | 506 ** | 120 ** | 0.24 ** | [56] |
| Flephedrone | 6.35 (4.2–9.5) * | >10 * | 5.8 (0.8–41) * | [9] | 12.5 (5.7–28) * | >33 * | na | [9] |
| 83.4 ** | 1290 ** | 15.4 ** | [56] | |||||
| 3–FMC | 1.7 (1.0–3.0) * | 56 (7–472) * | >10 * | [67] | ||||
| Pyrovalone | 0.035 (0.03–0.04) * | 13.0 (10.8–15.8) * | >100 * | [9] | ||||
| α-PVP | 0.2 (0.1–0.3) * | 237 (196–291) * | na | [65] | >100 * | >100 * | na | [55] |
| 0.04 (0.01–0.1) * | >100 * | >1000 * | [64] | |||||
| 12.8 ** | >10,000 ** | >781** | [56] | |||||
| α-PPP | 0.540 ± 0.076 * | 188 ± 12 * | na | [55] | >10 | >10 | na | [55] |
| 196 ** | >10,000 ** | >51 ** | [56] | |||||
| α-PVT | 0.342 ± 0.0049 * | 242 ± 41 * | na | [55] | ||||
| 3,4-DMMC | 9.4 (7.6–11.7) * | 1.1 (0.9–1.4) * | 0.12 (0.08–0.18) * | [66] | ||||
| Methylone | 4.82 (3.8–6.1) * | 15.5 (10–26) * | 3.3 (1.5–6.8) * | [9] | >100 * | >10 * | na | [9] |
| 2.0 (1.7–2.3) * | 68 (58–80) * | na | [65] | 133.0 ± 11.2 ** | 242.1 ± 48.3 ** | 1.82 ** | [8] | |
| 560 ± 50 ** | 230 ± 30 ** | na | [11] | |||||
| 12320 ± 133 ** | 1017 ± 59 ** | na | [10] | 117 ± 12 ** | 234 ± 35 ** | na | [10] | |
| Ethylone | 5.68 (4.9–6.5) * | 4.46 (3.8–5.2) * | 0.8 (0.6–1.1) * | [9] | >100 * | 9.9 (2.4–40) * | na | [9] |
| >10 * | 1.48 ± 0.25 * | na | [55] | |||||
| Butylone | 2.9 (2.5–3.4) * | 6.22 (4.3–9.0) * | 2.1 (1.3–3.6) * | [9] | >100 * | 5.5 (1.8–17) * | na | [9] |
| 1710 ± 320 ** | 680 ± 130 ** | na | [11] | |||||
| 400 ± 20 ** | 1430 ± 16 ** | na | [68] | (−) ** | 330 ± 40 ** | na | [68] | |
| Pentylone | 1.34 (1.0–1.7) * | 8.37 (5.4–13) * | 6.2 (3.2–13) * | [67] | >100 * | >100 * | na | [55] |
| 120 ± 10 ** | 1360 ± 100 ** | na | [68] | (−) ** | 1030 ± 180 ** | na | [68] | |
| MDPBP | 0.11 (0.07–0.16) * | 15 (5.4–39) * | 132 (34–557) * | [64] | >10 * | >10 * | na | [55] |
| MDPP | 1.08 ± 0.1 * | 126 ± 36 * | na | [69] | >10 * | >10 * | na | [55] |
| 0.53 (0.27–1.1) * | 75 (49–114) * | 141 (45–422) * | [64] | |||||
| MDPV | 0.031 (0.03–0.04) * | 9.3 (6.8–12.8) * | >100 * | [9] | >100 * | >100 * | na | [9] |
| 0.07 (0.07–0.08) * | 4.5 (4.0–5.2) * | na | [65] | |||||
| 0.05 (0.04–0.06) * | 9.6 (3.4–27) * | 192 (57–675) * | [64] | |||||
| 4.1 ± 0.5 ** | 3305 ± 305 ** | na | [10] | 2.3 ± 0.8 ** | (−) ** | na | [10] | |
| Compound | Monoamine Uptake Transporter Inhibition (IC50) | Monoamine Release (EC50) | ||||||
|---|---|---|---|---|---|---|---|---|
| DAT | SERT | DAT/SERT Ratio | Ref. | DAT | SERT | DAT/SERT Ratio | Ref. | |
| Amphetamine | 1.3 (0.83–2.0) * | >10 * | >10 * | [9] | 2.36 (1.7–3.3) * | >33 * | na | [9] |
| 1.3 (0.8–2.0) * | 45 (24–85) * | 35 (12–106) * | [55] | |||||
| 93 ± 17 ** | 3418 ± 314 ** | na | [10] | 5.8 ± 0.4 ** | 698 ± 71 ** | na | [10] | |
| Methamphetamine | 1.05 (0.75–1.5) * | >10 * | >10 * | [9,67] | 1.56 (0.9–2.8) * | >33 * | na | [9] |
| 1.1 (0.7–1.7) * | 18 (3–116) * | 17 (1.8–166) * | [64] | 0.435 ± 0.075 * | 23.3 ± 4.2 * | na | [55] | |
| 8.5 ± 1.4 ** | 1291 ± 241.6 ** | 152.0 ** | [8] | |||||
| Ephedrine | 46 (27–79) * | 230 (72–735) * | 5.0 (0.9–27) * | [64] | ||||
| MDMA | 17 (12–24) * | 1.36 (1.0–2.0) * | 0.08 (0.04–0.16) * | [9] | 22 (8.9–53) * | 5.63 (3.5–9.2) * | na | [9] |
| 31(8–118) * | 2.0 (1.4–3.0) * | 0.06 (0.01–0.4) * | [64] | 7.5 ± 2.3 * | 1.1 ± 0.29 * | na | [55] | |
| 51.2 ± 6.3 ** | 49.6 ± 5.4 ** | 0.97 ** | [8] | |||||
| MBDB | 22 (20–26) * | 2.04 (1.3–3.0) * | 0.09 (0.05–0.15) * | [9] | >100 * | 2.49 (1.0–6.9) * | na | [9] |
| MDEA | 9.3 (8.0–11) * | 1.27 (0.93–1.7) * | 0.14 (0.01–0.21) * | [9] | >100 * | 2.88 (1.6–5.0) * | na | [9] |
| Cocaine | 0.768 (0.6–1.0) * | 2.37 (2.0–2.9) * | 3.1 (2.0–4.8) * | [9] | >100 * | >100 * | na | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jîtcă, G.; Ősz, B.E.; Tero-Vescan, A.; Vari, C.E. Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review. Antioxidants 2021, 10, 381. https://doi.org/10.3390/antiox10030381
Jîtcă G, Ősz BE, Tero-Vescan A, Vari CE. Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review. Antioxidants. 2021; 10(3):381. https://doi.org/10.3390/antiox10030381
Chicago/Turabian StyleJîtcă, George, Bianca E. Ősz, Amelia Tero-Vescan, and Camil E. Vari. 2021. "Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review" Antioxidants 10, no. 3: 381. https://doi.org/10.3390/antiox10030381
APA StyleJîtcă, G., Ősz, B. E., Tero-Vescan, A., & Vari, C. E. (2021). Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review. Antioxidants, 10(3), 381. https://doi.org/10.3390/antiox10030381