
Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells



Abstract
:
{kind=link}
{kind=link}
{kind=link}
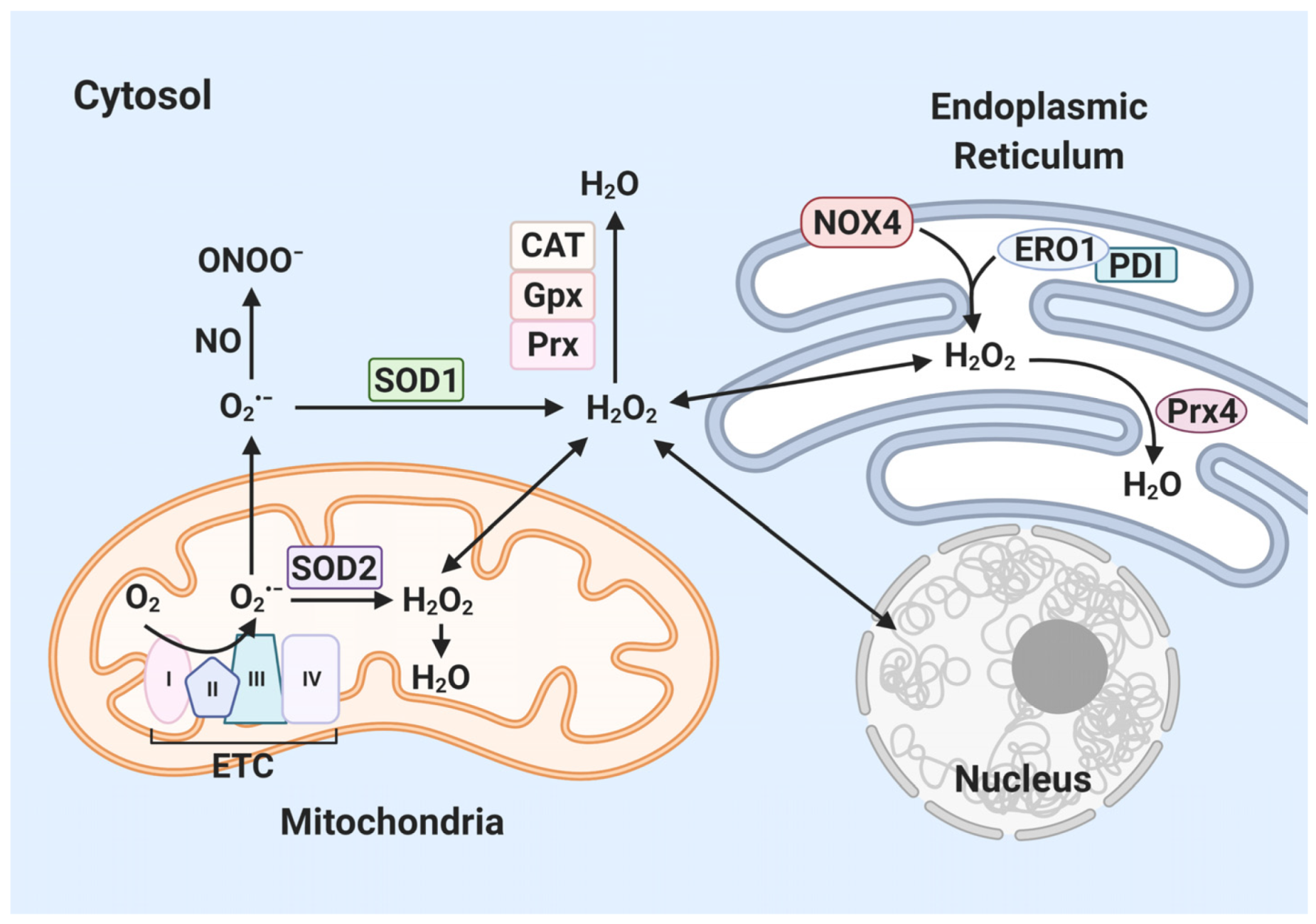{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Generation of ROS
2.1. ROS in Mitochondria
2.2. ROS in NADPH Oxidase
2.3. ROS in Endoplasmic Reticulum
3. Role of ROS in Innate Immunity
3.1. ROS in Innate Immunity
3.2. ROS in Bacterial Pathogenesis
3.3. ROS in Viral Pathogenesis
4. Oxidative Stress-Mediated ER Stress
5. Alteration of Mitochondrial Dynamics by Oxidative Stress
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From flies to men: ROS and the NADPH oxidase in phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Mayer, B.; Hemmens, B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem. Sci. 1997, 22, 477–481. [Google Scholar] [CrossRef]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Csányi, G.; Miller, F.J., Jr. Oxidative stress in cardiovascular disease. Int. J. Mol. Sci. 2014, 15, 6002–6008. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [Green Version]
- Thomson, N.C. Targeting oxidant-dependent mechanisms for the treatment of respiratory diseases and their comorbidities. Curr. Opin. Pharmacol. 2018, 40, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive oxygen production induced by the gut microbiota: Pharmacotherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Antimicrobial actions of reactive oxygen species. mBio 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatial properties of reactive oxygen species govern pathogen-specific immune system responses. Antioxid. Redox Signal. 2020, 32, 982–992. [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martínez de Marañón, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in Type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, G.; da Cruz, I.B.M.; González-Gallego, J. Chapter four—Manganese superoxide dismutase and oxidative stress modulation. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 68, pp. 87–130. [Google Scholar]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Cheng, M.L.; Ho, H.Y.; Chiu, D.T. The microbicidal and cytoregulatory roles of NADPH oxidases. Microbes Infect. 2011, 13, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Phagocyte-derived reactive species: Salvation or suicide? Trends Biochem. Sci. 2006, 31, 509–515. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2013, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, E.; Kastner, D.B.; Kaiser, C.A.; Fass, D. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 2004, 117, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Sevier, C.S.; Kaiser, C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 549. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, B.; Ferrari, D.M.; Klappa, P.; Pöhlmann, N.; Söling, H.D. Functional roles and efficiencies of the thioredoxin boxes of calcium-binding proteins 1 and 2 in protein folding. Biochem. J. 2001, 357, 83–95. [Google Scholar] [CrossRef]
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. EMBO Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.; Laurindo, F.R. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotugno, N.; Finocchi, A.; Cagigi, A.; Di Matteo, G.; Chiriaco, M.; Di Cesare, S.; Rossi, P.; Aiuti, A.; Palma, P.; Douagi, I. Defective B-cell proliferation and maintenance of long-term memory in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2015, 135, 753–761.e752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarsour, E.H.; Venkataraman, S.; Kalen, A.L.; Oberley, L.W.; Goswami, P.C. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell 2008, 7, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs. pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Akira, S. Identification and functions of pattern-recognition receptors. J. Allergy Clin. Immunol. 2010, 125, 985–992. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Celhar, T.; Magalhães, R.; Fairhurst, A.M. TLR7 and TLR9 in SLE: When sensing self goes wrong. Immunol. Res. 2012, 53, 58–77. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Bernard, J.J.; Cowing-Zitron, C.; Nakatsuji, T.; Muehleisen, B.; Muto, J.; Borkowski, A.W.; Martinez, L.; Greidinger, E.L.; Yu, B.D.; Gallo, R.L. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat. Med. 2012, 18, 1286–1290. [Google Scholar] [CrossRef]
- Laroux, F.S.; Romero, X.; Wetzler, L.; Engel, P.; Terhorst, C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J. Immunol. 2005, 175, 5596–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Frey, R.S.; Malik, A.B. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J. Clin. Investig. 2003, 112, 1234–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr. Opin. Hematol. 2002, 9, 11–17. [Google Scholar] [CrossRef]
- Takeya, R.; Sumimoto, H. Molecular mechanism for activation of superoxide-producing NADPH oxidases. Mol. Cells 2003, 16, 271–277. [Google Scholar]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Songhet, P.; Dumoutier, L.; Ferrero, I.; Janot, L.; Erard, F.; Bertout, J.; Leger, H.; et al. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127+ immune cells in spleen and mucosa. J. Immunol 2010, 185, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, T.; Kuwano, Y.; Teshima-Kondo, S.; Takeya, R.; Sumimoto, H.; Kishi, K.; Tsunawaki, S.; Hirayama, T.; Rokutan, K. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J. Immunol. 2004, 172, 3051–3058. [Google Scholar] [CrossRef]
- Joo, J.H.; Ryu, J.H.; Kim, C.H.; Kim, H.J.; Suh, M.S.; Kim, J.O.; Chung, S.Y.; Lee, S.N.; Kim, H.M.; Bae, Y.S.; et al. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid. Redox Signal. 2012, 16, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Makni-Maalej, K.; Boussetta, T.; Hurtado-Nedelec, M.; Belambri, S.A.; Gougerot-Pocidalo, M.A.; El-Benna, J. The TLR7/8 agonist CL097 primes N-formyl-methionyl-leucyl-phenylalanine-stimulated NADPH oxidase activation in human neutrophils: Critical role of p47phox phosphorylation and the proline isomerase Pin1. J. Immunol. 2012, 189, 4657–4665. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-G.; Lee, S.-H.; Park, D.-W.; Lee, S.-H.; Yoon, H.-S.; Chin, B.-R.; Kim, J.-H.; Kim, J.-R.; Baek, S.-H. Toll-like receptor 9-stimulated monocyte chemoattractant protein-1 is mediated via JNK-cytosolic phospholipase A2-ROS signaling. Cell. Signal. 2008, 20, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Broz, P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. 2017, 38, 744–757. [Google Scholar] [CrossRef]
- Park, J.-H.; Kim, Y.-G.; McDonald, C.; Kanneganti, T.-D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Núñez, G. RICK/RIP2 Mediates Innate Immune Responses Induced through Nod1 and Nod2 but Not TLRs. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, T.; Kaneko, T.; Yoshimura, A.; Silverman, N.; Hara, Y. NOD1 and NOD2 mediate sensing of periodontal pathogens. J. Dent. Res. 2010, 89, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, S.; Till, A.; Sina, C.; Arlt, A.; Grasberger, H.; Schreiber, S.; Rosenstiel, P. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 2009, 122, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohchi, C.; Inagawa, H.; Nishizawa, T.; Soma, G.-I. ROS and innate immunity. Anticancer Res. 2009, 29, 817–821. [Google Scholar] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2017, 162, 603–610. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Gardina, P.; Myers, T.G.; Leto, T.L. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucosal. Immunol. 2011, 4, 158–171. [Google Scholar] [CrossRef]
- Brandes, R.P.; Viedt, C.; Nguyen, K.; Beer, S.; Kreuzer, J.; Busse, R.; Görlach, A. Thrombin-induced MCP-1 expression involves activation of the p22phox-containing NADPH oxidase in human vascular smooth muscle cells. Thromb. Haemost. 2001, 85, 1104–1110. [Google Scholar] [PubMed]
- Cho, S.O.; Lim, J.W.; Kim, H. Red ginseng extract inhibits the expression of MCP-1 and iNOS in Helicobacter pylori-infected gastric epithelial cells by suppressing the activation of NADPH oxidase and Jak2/Stat3. J. Ethnopharmacol. 2013, 150, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Batra, S.; Lira, S.A.; Kolls, J.K.; Jeyaseelan, S. CXCL1 regulates pulmonary host defense to Klebsiella infection via CXCL2, CXCL5, NF-κB, and MAPKs. J. Immunol. 2010, 185, 6214–6225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaddi, K.; Newton, R.C. Comparison of biological responses of human monocytes and THP-1 cells to chemokines of the intercrine-β family. J. Leukoc. Biol. 1994, 55, 756–762. [Google Scholar] [CrossRef]
- Furie, M.B.; Randolph, G.J. Chemokines and tissue injury. Am. J. Pathol. 1995, 146, 1287–1301. [Google Scholar]
- Tan, J.H.; Ludeman, J.P.; Wedderburn, J.; Canals, M.; Hall, P.; Butler, S.J.; Taleski, D.; Christopoulos, A.; Hickey, M.J.; Payne, R.J.; et al. Tyrosine sulfation of chemokine receptor CCR2 enhances interactions with both monomeric and dimeric forms of the chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 2013, 288, 10024–10034. [Google Scholar] [CrossRef] [Green Version]
- Shekhova, E. Mitochondrial reactive oxygen species as major effectors of antimicrobial immunity. PLoS Pathog. 2020, 16, e1008470. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, W.; Domann, E.; Chakraborty, T.; Mannala, G.; Lips, K.S.; Heiss, C.; Schnettler, R.; Alt, V. TLR9 mediates S. aureus killing inside osteoblasts via induction of oxidative stress. BMC Microbiol. 2016, 16, 230. [Google Scholar] [CrossRef] [Green Version]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.e625. [Google Scholar] [CrossRef] [Green Version]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acín-Pérez, R.; Carrascoso, I.; Baixauli, F.; Roche-Molina, M.; Latorre-Pellicer, A.; Fernández-Silva, P.; Mittelbrunn, M.; Sanchez-Madrid, F.; Pérez-Martos, A.; Lowell, C.A.; et al. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab. 2014, 19, 1020–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling. Am. J. Respir. Crit. Care Med. 2002, 166, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Hohn, D.C.; Lehrer, R.I. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J. Clin. Investig. 1975, 55, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, J.; Wolach, O.; Gavrieli, R.; Wolach, B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti Infect. 2012, 10, 881–894. [Google Scholar] [CrossRef]
- Myers, J.T.; Tsang, A.W.; Swanson, J.A. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J. Immunol. 2003, 171, 5447–5453. [Google Scholar] [CrossRef] [Green Version]
- KuoLee, R.; Harris, G.; Conlan, J.W.; Chen, W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 2011, 13, 447–456. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Redmond, S.; Jones, A.A.; Ramakrishnan, L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell 2019, 178, 1344–1361.e1311. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, J.; Laganière, J.; Mehl, I.R.; Barish, G.D.; Chong, L.W.; Li, X.; Scheffler, I.E.; Mock, D.C.; Bataille, A.R.; Robert, F.; et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev. 2007, 21, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Flory, E.; Kunz, M.; Scheller, C.; Jassoy, C.; Stauber, R.; Rapp, U.R.; Ludwig, S. Influenza virus-induced NF-kappaB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IkappaB kinase. J. Biol. Chem. 2000, 275, 8307–8314. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.J.; Liao, C.L.; Lin, Y.L. Replication-incompetent virions of Japanese encephalitis virus trigger neuronal cell death by oxidative stress in a culture system. J. Gen. Virol. 2004, 85, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Hooper, L.C.; Okuno, T.; Takada, Y.; Hooks, J.J. Inhibition of HSV-1 by chemoattracted neutrophils: Supernatants of corneal epithelial cells (HCE) and macrophages (THP-1) treated with virus components chemoattract neutrophils (PMN), and supernatants of PMN treated with these conditioned media inhibit viral growth. Arch. Virol. 2012, 157, 1377–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglié, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef] [Green Version]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Niu, J.; Wu, S.; Chen, M.; Xu, K.; Guo, Q.; Lu, A.; Zhao, L.; Sun, B.; Meng, G. Hyperactivation of the NLRP3 inflammasome protects mice against influenza A virus infection via IL-1β mediated neutrophil recruitment. Cytokine 2019, 120, 115–124. [Google Scholar] [CrossRef]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J. Virol. 2011, 85, 12585–12593. [Google Scholar] [CrossRef] [Green Version]
- Speir, E.; Shibutani, T.; Yu, Z.X.; Ferrans, V.; Epstein, S.E. Role of reactive oxygen intermediates in cytomegalovirus gene expression and in the response of human smooth muscle cells to viral infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef]
- Flores, S.C.; Marecki, J.C.; Harper, K.P.; Bose, S.K.; Nelson, S.K.; McCord, J.M. Tat protein of human immunodeficiency virus type 1 represses expression of manganese superoxide dismutase in HeLa cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Guillin, O.M.; Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium, selenoproteins and viral infection. Nutrients 2019, 11, 2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.-M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Béraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef] [Green Version]
- De Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Snelgrove, R.J.; Edwards, L.; Rae, A.J.; Hussell, T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006, 36, 1364–1373. [Google Scholar] [CrossRef]
- Snelgrove, R.; Williams, A.; Thorpe, C.; Hussell, T. Manipulation of immunity to and pathology of respiratory infections. Expert Rev. Anti Infect. 2004, 2, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Ryan, L.K.; Bishop, L.; Folz, R.J. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L69–L78. [Google Scholar] [CrossRef]
- Vlahos, R.; Stambas, J.; Selemidis, S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharm. Sci. 2012, 33, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T. Role of free radicals in viral pathogenesis and mutation. Rev. Med. Virol. 2001, 11, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [Green Version]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of oxidative stress and mitochondrial dysfunction in sepsis and potential therapies. Oxid. Med. Cell Longev. 2017, 2017, 5985209. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Hartmann, M. Mechanisms of hemolysis during sepsis. Inflammation 2018, 41, 1569–1581. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Diotallevi, A.; Magnani, M. Endoplasmic reticulum stress and unfolded protein response in infection by intracellular parasites. Future Sci. OA 2017, 3, FSO198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-A.; Song, C.-H. Insights into the role of endoplasmic reticulum stress in infectious diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.-J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma—From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10, eaal2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [Green Version]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arter. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Lardy, B.; Krause, K.-H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef]
- Santos, C.X.; Stolf, B.S.; Takemoto, P.V.; Amanso, A.M.; Lopes, L.R.; Souza, E.B.; Goto, H.; Laurindo, F.R. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J. Leukoc. Biol. 2009, 86, 989–998. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Palejwala, A.; Tepikin, A.V.; Petersen, O.H.; Watson, A.J. Menadione-induced apoptosis: Roles of cytosolic Ca2+ elevations and the mitochondrial permeability transition pore. J. Cell Sci. 2002, 115, 485–497. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.-P.; De Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 1612–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, D.L. Redox regulation of endothelial canonical transient receptor potential channels. Antioxid. Redox Signal. 2011, 15, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Pillich, H.; Loose, M.; Zimmer, K.-P.; Chakraborty, T. Diverse roles of endoplasmic reticulum stress sensors in bacterial infection. Mol. Cell Pediatr. 2016, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Bischof, L.J.; Kao, C.Y.; Los, F.C.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef]
- Loose, M.; Hudel, M.; Zimmer, K.-P.; Garcia, E.; Hammerschmidt, S.; Lucas, R.; Chakraborty, T.; Pillich, H. Pneumococcal hydrogen peroxide-induced stress signaling regulates inflammatory genes. J. Infect. Dis. 2015, 211, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Vasallo, C.; Gastaminza, P. Cellular stress responses in hepatitis C virus infection: Mastering a two-edged sword. Virus Res. 2015, 209, 100–117. [Google Scholar] [CrossRef]
- Ríos-Ocampo, W.A.; Navas, M.C.; Faber, K.N.; Daemen, T.; Moshage, H. The cellular stress response in hepatitis C virus infection: A balancing act to promote viral persistence and host cell survival. Virus Res. 2019, 263, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ocampo, W.A.; Navas, M.C.; Buist-Homan, M.; Faber, K.N.; Daemen, T.; Moshage, H. Hepatitis C Virus Proteins Core and NS5A Are Highly Sensitive to Oxidative Stress-Induced Degradation after eIF2α/ATF4 Pathway Activation. Viruses 2020, 12, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.-C.; Lai, C.-C.; Shiu, S.-L.; Chuang, P.-H.; Tzou, B.-C.; Lin, Y.-Y.; Tsai, F.-J.; Lin, C.-W. Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect. 2010, 12, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.; Wang, L.; Graczyk, T.M.; Block, T.M.; Romano, P.R. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J. Virol. 2002, 76, 9588–9599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuaita, B.H.; Burkholder, K.M.; Boles, B.R.; O’Riordan, M.X. The Endoplasmic Reticulum Stress Sensor Inositol-Requiring Enzyme 1α Augments Bacterial Killing through Sustained Oxidant Production. mBio 2015, 6, e00705–e00715. [Google Scholar] [CrossRef] [Green Version]
- Celli, J.; Tsolis, R.M. Bacteria, the endoplasmic reticulum and the unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 2015, 13, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Khan, M.; Magnani, D.D.; Harms, J.S.; Durward, M.; Radhakrishnan, G.K.; Liu, Y.P.; Splitter, G.A. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog. 2013, 9, e1003785. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.F.; Starr, T.; Winter, M.G.; den Hartigh, A.B.; Child, R.; Knodler, L.A.; van Dijl, J.M.; Celli, J.; Tsolis, R.M. Sensing of bacterial type IV secretion via the unfolded protein response. mBio 2013, 4, e00418-12. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Shin, D.M.; Kang, G.; Kim, K.H.; Park, J.B.; Hur, G.M.; Lee, H.M.; Lim, Y.J.; Park, J.K.; Jo, E.K.; et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 2010, 584, 2445–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.J.; Choi, J.A.; Lee, J.H.; Choi, C.H.; Kim, H.J.; Song, C.H. Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis 2015, 20, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kolattukudy, P.E. Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell Signal. 2012, 24, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-J.; Choi, J.-A.; Choi, H.-H.; Cho, S.-N.; Kim, H.-J.; Jo, E.-K.; Park, J.-K.; Song, C.-H. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e28531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.-Y.; Lim, Y.-J.; Choi, J.-A.; Lee, J.-H.; Jo, S.-H.; Oh, S.-M.; Song, C.-H. The role of prostate apoptosis response-4 (Par-4) in Mycobacterium tuberculosis infected macrophages. Sci. Rep. 2016, 6, 32079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.H.; Choi, J.-A.; Lim, Y.-J.; Lee, J.; Cho, S.-N.; Oh, S.-M.; Go, D.; Kim, S.-H.; Song, C.-H. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget 2017, 8, 58686–58698. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.-J.; Choi, H.-H.; Choi, J.-A.; Jeong, J.A.; Cho, S.-N.; Lee, J.-H.; Park, J.B.; Kim, H.-J.; Song, C.-H. Mycobacterium kansasii-induced death of murine macrophages involves endoplasmic reticulum stress responses mediated by reactive oxygen species generation or calpain activation. Apoptosis 2013, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-M.; Lim, Y.-J.; Choi, J.-A.; Lee, J.; Cho, S.-N.; Go, D.; Kim, S.-H.; Song, C.-H. TNF-α-mediated ER stress causes elimination of Mycobacterium fortuitum reservoirs by macrophage apoptosis. FASEB J. 2018, 32, 3993–4003. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Cho, S.-N.; Lim, Y.-J.; Choi, J.-A.; Lee, J.; Go, D.; Song, C.-H. Phagocytosis influences the intracellular survival of Mycobacterium smegmatis via the endoplasmic reticulum stress response. Cell Biosci. 2018, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Go, D.; Lee, J.; Choi, J.-A.; Cho, S.-N.; Kim, S.-H.; Son, S.-H.; Song, C.-H. Reactive oxygen species-mediated endoplasmic reticulum stress response induces apoptosis of Mycobacterium avium-infected macrophages by activating regulated IRE1-dependent decay pathway. Cell Microbiol. 2019, 21, e13094. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Sabouny, R.; Shutt, T.E. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem. Sci. 2020, 45, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Chakrabarti, O. Regulation of Mitofusin1 by Mahogunin Ring Finger-1 and the proteasome modulates mitochondrial fusion. Biochim. Biophys. Acta 2016, 1863, 3065–3083. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef]
- Pendin, D.; Filadi, R.; Pizzo, P. The concerted action of mitochondrial dynamics and positioning: New characters in cancer onset and progression. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Nagdas, S.; Kashatus, D.F. The interplay between oncogenic signaling networks and mitochondrial dynamics. Antioxidants. 2017, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol. 2007, 17, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Youle, R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003, 10, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Fielden, L.F.; Kang, Y.; Newton, H.J.; Stojanovski, D. Targeting mitochondria: How intravacuolar bacterial pathogens manipulate mitochondria. Cell Tissue Res. 2017, 367, 141–154. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.H.; Yates, C.; You, Z.; Tan, M. Diverse roles of mitochondria in immune responses: Novel insights into immuno-metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Okuda, M.; Otani, K.; Wang, T.; Li, Y.; Weinman, S.A. Mitochondrial dysfunction in hepatitis C. J. Clin. Gastroenterol. 2005, 39, S162–S166. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.-M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Meng, G.; Jiang, A.; Chen, A.; Dahlhaus, M.; Gonzalez, P.; Beltinger, C.; Wei, J. Mitophagy switches cell death from apoptosis to necrosis in NSCLC cells treated with oncolytic measles virus. Oncotarget 2014, 5, 3907–3918. [Google Scholar] [CrossRef]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef] [Green Version]
- Mohasin, M.; Balbirnie-Cumming, K.; Fisk, E.; Prestwich, E.C.; Russell, C.D.; Marshall, J.; Pridans, C.; Allen, S.P.; Shaw, P.J.; De Vos, K.J.; et al. Macrophages utilize mitochondrial fission to enhance mROS production during responses to Streptococcus pneumoniae. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Luo, Z.-Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.; Kuang, L.; Xiang, X.; Zhang, J.; Zhu, Y.; Wu, Y.; Yan, Q.; Liu, L.; Li, T. Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis. 2020, 11, 251. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.-J.; Lee, J.; Oh, S.J.; Yoon, M.-S.; Jang, S.-S.; Holland, R.L.; Reno, M.L.; Hamad, M.N.; Maeda, T.; Chung, H.J.; et al. Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593.e588. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, L.D.; Pypaert, M.; Flavell, R.A.; Galán, J.E. A Salmonella protein causes macrophage cell death by inducing autophagy. J. Cell Biol. 2003, 163, 1123–1131. [Google Scholar] [CrossRef]
- Lobet, E.; Willemart, K.; Ninane, N.; Demazy, C.; Sedzicki, J.; Lelubre, C.; De Bolle, X.; Renard, P.; Raes, M.; Dehio, C.; et al. Mitochondrial fragmentation affects neither the sensitivity to TNFα-induced apoptosis of Brucella-infected cells nor the intracellular replication of the bacteria. Sci. Rep. 2018, 8, 5173. [Google Scholar] [CrossRef] [Green Version]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Escoll, P.; Song, O.-R.; Viana, F.; Steiner, B.; Lagache, T.; Olivo-Marin, J.-C.; Impens, F.; Brodin, P.; Hilbi, H.; Buchrieser, C. Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe 2017, 22, 302–316.e307. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.R.; Reimer, A.; Sharan, M.; Kozjak-Pavlovic, V.; Eulalio, A.; Prusty, B.K.; Fraunholz, M.; Karunakaran, K.; Rudel, T. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J. Cell Biol. 2017, 216, 1071–1089. [Google Scholar] [CrossRef] [PubMed]
- Asalla, S.; Mohareer, K.; Banerjee, S. Small molecule mediated restoration of mitochondrial function augments anti-mycobacterial activity of human macrophages subjected to cholesterol induced asymptomatic dyslipidemia. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamwal, S.; Midha, M.K.; Verma, H.N.; Basu, A.; Rao, K.V.S.; Manivel, V. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci. Rep. 2013, 3, 1328. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Choi, J.A.; Cho, S.N.; Son, S.H.; Song, C.H. Mitofusin 2-deficiency suppresses Mycobacterium tuberculosis survival in macrophages. Cells 2019, 8, 1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Goh, J.-Y.; Xiao, L.; Xian, H.; Lim, K.-L.; Liou, Y.-C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Song, C.-H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. https://doi.org/10.3390/antiox10060872
Lee J, Song C-H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants. 2021; 10(6):872. https://doi.org/10.3390/antiox10060872
Chicago/Turabian StyleLee, Junghwan, and Chang-Hwa Song. 2021. "Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells" Antioxidants 10, no. 6: 872. https://doi.org/10.3390/antiox10060872
APA StyleLee, J., & Song, C. -H. (2021). Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants, 10(6), 872. https://doi.org/10.3390/antiox10060872