
Immunological Aspects of X-Linked Chronic Granulomatous Disease Female Carriers

Abstract
:1. Introduction
2. Material and Methods
2.1. Patients and Informed Consent
2.2. Clinical Features
2.3. Molecular and Biochemical Investigations
2.4. Flow-Cytometry Studies
2.5. Statistical Analysis
3. Results
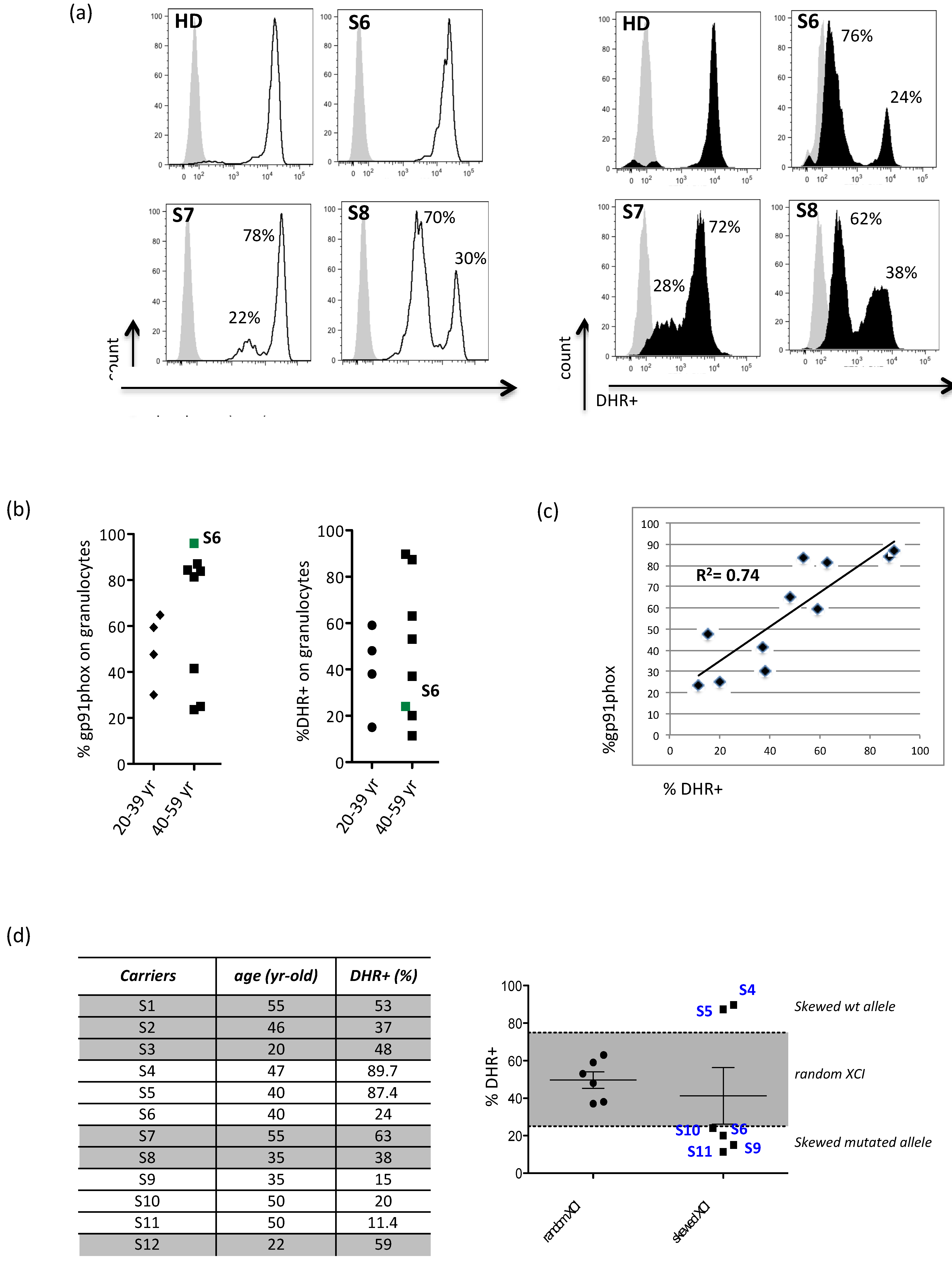
3.1. Evidence for X-Chromosome Inactivation Age-Independent in X-CGD Carriers
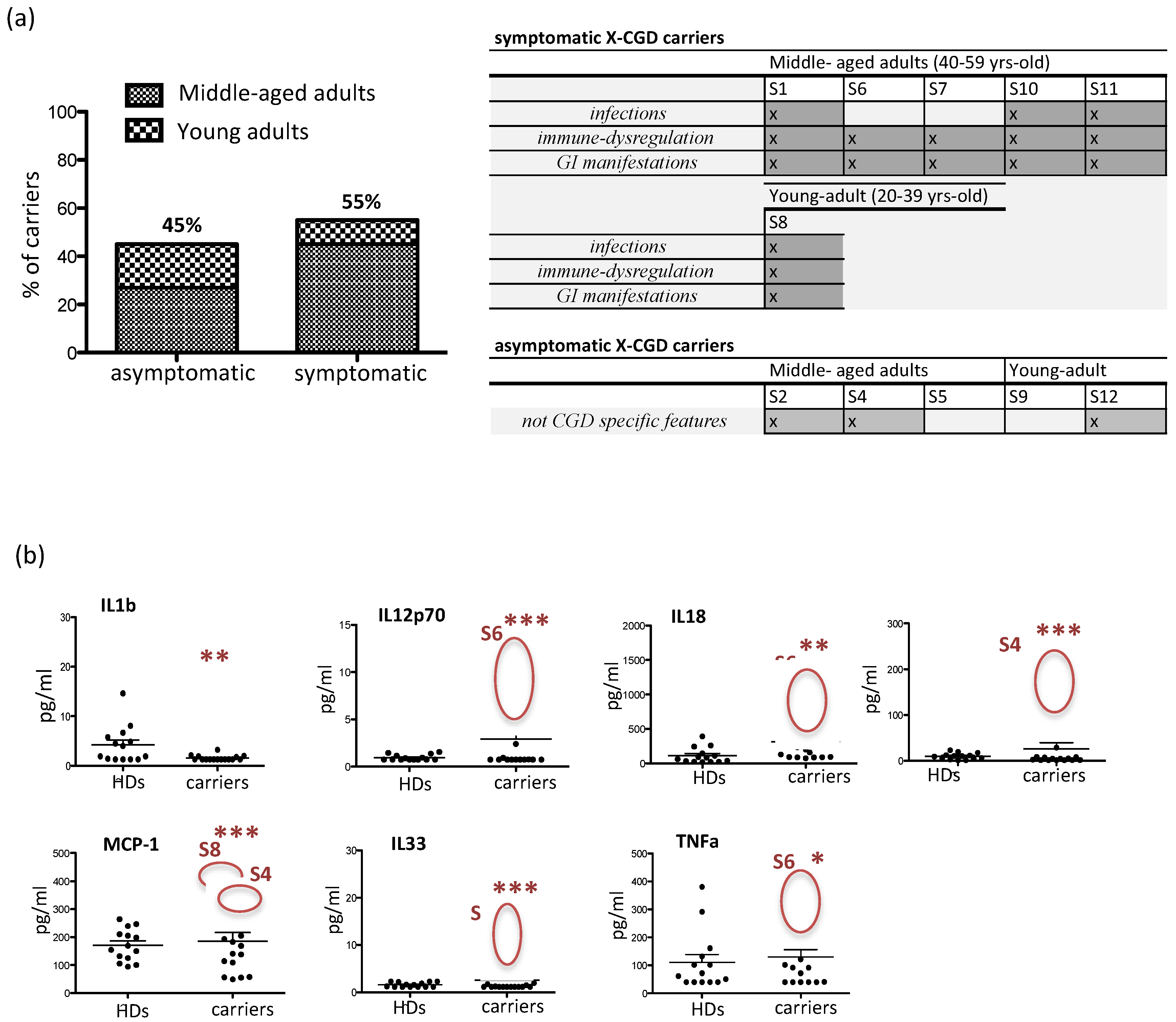
3.2. Clinical Manifestations and Inflammatory Cytokine/Chemokine Profiling in X-CGD Carriers
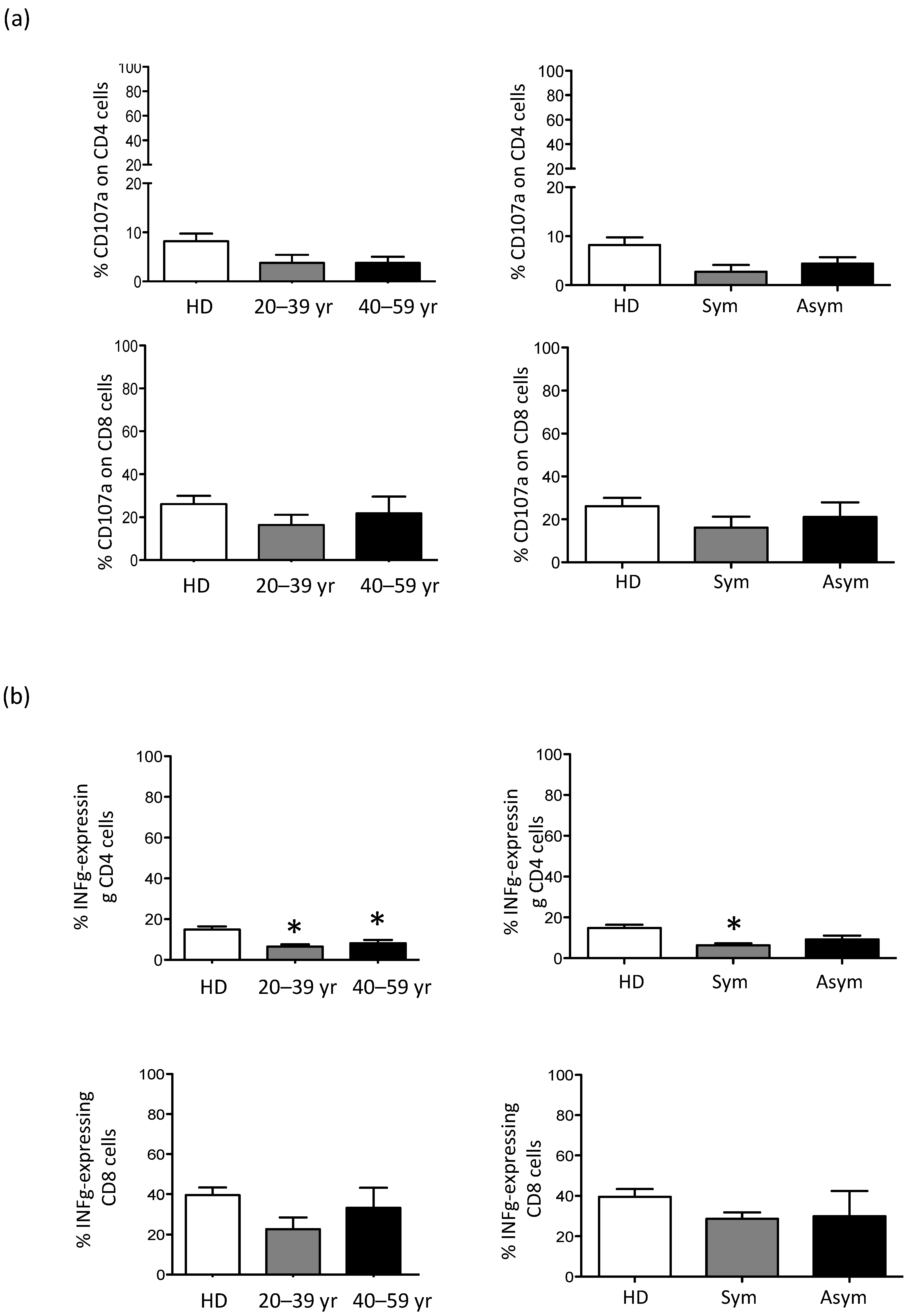
3.3. CD56bright NK Cell Are Expanded in Older X-CGD Carriers
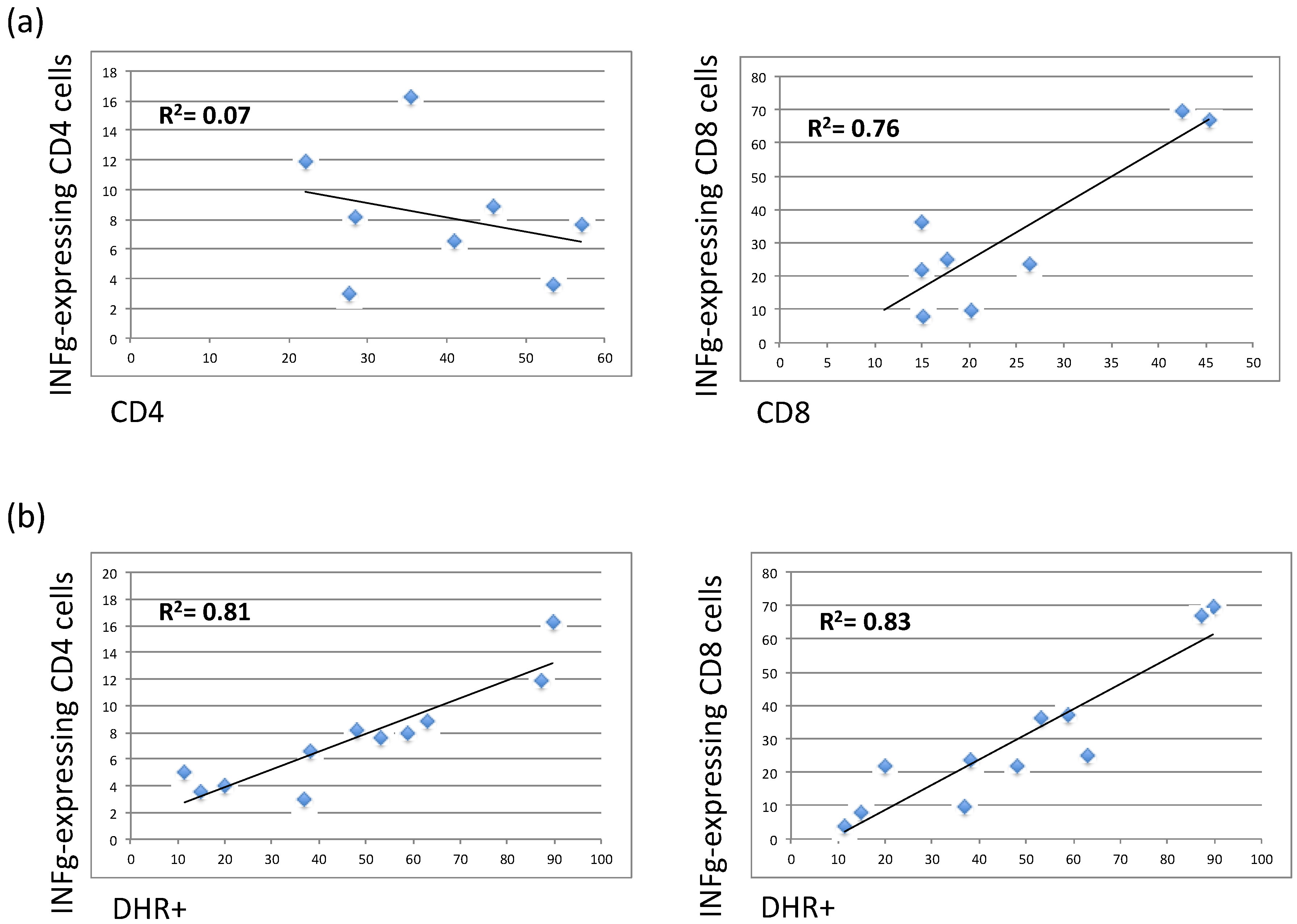
3.4. The Residual Production of ROS (% DHR+ Cells) Correlates in X-CGD Carriers with the Reduced Percentage of Both CD8 Cells and INFg-CD4 Expressing Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Chronic granulomatous disease (CGD) |
| Nicotinamide dinucleotide phosphate (NADPH) |
| X-linked CGD (X-CGD) |
| Reactive oxygen species (ROSs) |
| Inflammatory bowel disease (IBD) |
| X-chromosome inactivation (XCI) |
| Dihydrorhodamine 123 (DHR) |
| Peripheral blood mononuclear cell (PBMC) |
| Phorbol 12-myristate 13-acetate (PMA) |
| Healthy Donor (HD) |
| Wild type (wt) |
References
- Chiriaco, M.; Salfa, I.; Di Matteo, G.; Rossi, P.; Finocchi, A. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr. Allergy Immunol. 2016, 27, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Arnadottir, G.A.; Norddahl, G.L.; Gudmundsdottir, S.; Agustsdottir, A.B.; Sigurdsson, S.; Jensson, B.O.; Bjarnadottir, K.; Theodors, F.; Benonisdottir, S.; Ivarsdottir, E.V.; et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. 2018, 9, 4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, N.L.; Jameson, M.B.; Creech, C.B. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S2–S5. [Google Scholar] [CrossRef] [Green Version]
- De Ravin, S.S.; Naumann, N.; Cowen, E.W.; Friend, J.; Hilligoss, D.; Marquesen, M.; Balow, J.E.; Barron, K.S.; Turner, M.L.; Gallin, J.I.; et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J. Allergy Clin. Immunol. 2008, 122, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Crockard, A.D.; Thompson, J.M.; Boyd, N.A.; Haughton, D.J.; McCluskey, D.R.; Turner, C.P. Diagnosis and Carrier Detection of Chronic Granulomatous Disease in Five Families by Flow Cytometry. Int. Arch. Allergy Immunol. 1997, 114, 144–152. [Google Scholar] [CrossRef]
- Chiriaco, M.; Di Matteo, G.; Sinibaldi, C.; Giardina, E.; Nardone, A.M.; Folgori, L.; D’Argenio, P.; Rossi, P.; Finocchi, A. Identification of Deletion Carriers in X-Linked Chronic Granulomatous Disease by Real-Time PCR Genetic Testing and Molecular Biomarkers. Genet. Test. Mol. Biomark. 2009, 13, 785–789. [Google Scholar] [CrossRef]
- Hatakeyama, C.; Anderson, C.L.; Beever, C.L.; Penaherrera, M.S.; Brown, C.J.; Robinson, W.P. The dynamics of X-inactivation skewing as women age. Clin. Genet. 2004, 66, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Battersby, A.C.; Cale, C.M.; Goldblatt, D.; Gennery, A.R. Clinical Manifestations of Disease in X-Linked Carriers of Chronic Granulomatous Disease. J. Clin. Immunol. 2013, 33, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Hauck, F.; Koletzko, S.; Walz, C.; Von Bernuth, H.; Klenk, A.; Schmid, I.; Belohradsky, B.H.; Klein, C.; Bufler, P.; Albert, M.H. Diagnostic and Treatment Options for Severe IBD in Female X-CGD Carriers with Non-random X-inactivation. J. Crohns Colitis 2016, 10, 112–115. [Google Scholar] [CrossRef]
- Marciano, B.E.; Zerbe, C.S.; Falcone, E.L.; Ding, L.; DeRavin, S.S.; Daub, J.; Kreuzburg, S.; Yockey, L.; Hunsberger, S.; Foruraghi, L.; et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 2018, 141, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koker, M.Y.; Sanal, O.; De Boer, M.; Tezcan, I.; Metin, A.; Tan, C.; Ersoy, F.; Roos, D. Skewing of X-chromosome inactivation in three generations of carriers with X-linked chronic granulomatous disease within one family. Eur. J. Clin. Investig. 2006, 36, 257–264. [Google Scholar] [CrossRef]
- Battersby, A.C.; Braggins, H.; Pearce, M.S.; Cale, C.M.; Burns, S.O.; Hackett, S.; Hughes, S.; Barge, D.; Goldblatt, D.; Gennery, A.R. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J. Allergy Clin. Immunol. 2017, 140, 628–630. [Google Scholar] [CrossRef] [Green Version]
- Di Matteo, G.; Giordani, L.; Finocchi, A.; Ventura, A.; Chiriaco, M.; Blancato, J.; Sinibaldi, C.; Plebani, A.; Soresina, A.; Pignata, C.; et al. Molecular characterization of a large cohort of patients with Chronic Granulomatous Disease and identification of novel CYBB mutations: An Italian multicenter study. Mol. Immunol. 2009, 46, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Chiriaco, M.; Casciano, F.; Di Matteo, G.; Gentner, B.; Claps, A.; Di Cesare, S.; Cotugno, N.; D’Argenio, P.; Rossi, P.; Aiuti, A.; et al. Impaired X-CGD T cell compartment is gp91phox-NADPH oxidase independent. Clin. Immunol. 2018, 193, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D.G. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 1996, 88, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, D.; van Leeuwen, K.; Amy; Hsu, P.; Priel, D.L.; Begtrup, A.; Brandon, R.; Stasia, M.J.; Bakri, F.G.; Köker, N.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (fourth update). Blood Cells Mol. Dis. 2021. under review. [Google Scholar] [CrossRef] [Green Version]
- Puck, J.M.; Willard, H.F. X Inactivation in Females with X-Linked Disease. N. Engl. J. Med. 1998, 338, 325–328. [Google Scholar] [CrossRef]
- Veyver, I.B.V.D. Skewed X Inactivation in X-Linked Disorders. Semin. Reprod. Med. 2001, 19, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Cotugno, N.; Finocchi, A.; Cagigi, A.; Di Matteo, G.; Chiriaco, M.; Di Cesare, S.; Rossi, P.; Aiuti, A.; Palma, P.; Douagi, I. Defective B-cell proliferation and maintenance of long-term memory in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2015, 135, 753–761.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.E.; Fielding, A.K.; Harrison, C.N.; Linch, D.C. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br. J. Haematol. 1997, 98, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Rösen-Wolff, A.; Soldan, W.; Heyne, K.; Bickhardt, J.; Gahr, M.; Roesler, J. Increased susceptibility of a carrier of X-linked chronic granulomatous disease (CGD) to Aspergillus fumigatus infection associated with age-related skewing of lyonization. Ann. Hematol. 2001, 80, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Chen, Y.C.; Lee, W.I.; Huang, J.L.; Chen, L.C.; Ou, L.S.; Yao, T.C.; Jaing, T.H. Clinical Features of Female Taiwanese Carriers with X-linked Chronic Granulomatous Disease from 2004 to 2019. J. Clin. Immunol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Schumann, M.; Kühnel, A. Pathophysiological Role of TNF in Inflammatory Bowel Disease:Anti-Tumor Necrosis Factor Therapy in Inflammatory Bowel Disease. Front. Gastrointest. Res. 2015, 34, 49–55. [Google Scholar]
- Feldman, A.M.; Combes, A.; Wagner, D.; Kadakomi, T.; Kubota, T.; Li, Y.Y.; McTiernan, C. The role of tumor necrosis factor in the pathophysiology of heart failure. J. Am. Coll. Cardiol. 2000, 35, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M. Role of IL-33 in inflammation and disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Saini, V.; Arora, S. MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 2010, 411, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Sedimbi, S.K.; Hägglöf, T.; Karlsson, M.C. IL-18 in inflammatory and autoimmune disease. Cell. Mol. Life Sci. 2013, 70, 4795–4808. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Ni, D.; Brissette, R.; Chou, M.; Hussain, M.; Gill, D.S.; Liao, M.J.; Testa, D. Interferon-alpha 2 variants in the human genome. J. Interferon Cytokine Res. 1995, 15, 341–349. [Google Scholar] [CrossRef]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56bright NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Aging of the Immune System. Mechanisms and Therapeutic Targets. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S5), S422–S428. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, X.; Saredy, J.; Yuan, Z.; Yang, X.; Wang, H. Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol. 2020, 37, 101759. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Female Analysed (Range of Age) | Mutation in CYBB Gene | Protein Expression (wt/mut) | X-Linked Carrier Status |
|---|---|---|---|
| S1 (B) | 581bp del | X91/ X91° | carrier |
| S2 (B) | c.925G>A/p.E309K | X91/X91° | carrier |
| S3 (A) | c.469C>T /p.R157X. | X91/X91° | carrier |
| S4 (B) | c.252G>A/p.A84 | X91/X91° | carrier |
| S5 (B) | c.742dupA p.Ile248AsnFsX36 | X91/X91° | carrier |
| S6 (B) | c.1531T>G /p.Y511D | X91/X91+ | carrier |
| S7 (B) | 1637 Mbp del + McLeod | X91/X91° | carrier |
| S8 (A) | 32,72 Kb del | X91/X91° | carrier |
| S9 (A) | c.736C>T/p.Q246X | X91/X91° | carrier |
| S10 (B) | c.1357T>A/p.W453R | X91/X91° | carrier |
| S11 (B) | ex9-11del | X91/X91° | carrier |
| S12 (A) | 1637 Mbp del + McLeod | X91/X91° | carrier |
| S13 | - | X91/X91 | HD |
| S14 | - | X91/X91 | HD |
| S15 | - | X91/X91 | HD |
| HDs (n = 25) | - | X91/X91 | HD |
| HD | Carriers 20–39 yr | Carriers 40–59 yr | |
|---|---|---|---|
| gated on lymphocytes | (%) | (%) | (%) |
| T cells (CD3+) | 72.6 ± 1.9 | 72 ± 3.4 | 63 ± 8 |
| CD4 Tcells (CD3+CD4+) | 45.7 ± 3.1 | 44 ± 5.6 | 41 ±5.3 |
| CD8 T cells (CD3+CD8+) | 26.5 ± 1.4 | 19 ± 2.6 | 28 ± 6.1 |
| B cells (CD19+) | 8.3 ± 1 | 12.6 ± 0.4 | 15.7 ± 2.7 ** |
| NKT cells (CD3+CD56+) | 4.8 ± 0.6 | 4.3 ± 0.4 | 8.3 ± 2.7 |
| NK cells (CD3-CD56+) | 9.3 ± 1.4 | 5.8 ± 0.3 | 6.8 ± 1.3 |
| gated on NK cells | |||
| NK bright (CD16-) | 17.6 ± 1.9 | 20 ± 8.3 | 34 ± 6.2 ** |
| NK dim (CD16+) | 74.5 ± 3.14 | 68 ± 4.8 | 61 ± 4 |
| CD16+CD14- (non-classical) | 11.5 ± 2.1 | nd | 8.4 ±3.1 |
| CD16+CD14low (late-intermediate) | 0.6 ± 0.19 | nd | 1.2 ± 0.4 |
| CD16+CD14bright (early-intermediate) | 0.7 ± 0.22 | nd | 0.7 ± 0.1 |
| CD16-CD14+ (classical) | 17.6 ± 2.6 | nd | 17.71 ± 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiriaco, M.; Salfa, I.; Ursu, G.M.; Cifaldi, C.; Di Cesare, S.; Rossi, P.; Di Matteo, G.; Finocchi, A. Immunological Aspects of X-Linked Chronic Granulomatous Disease Female Carriers. Antioxidants 2021, 10, 891. https://doi.org/10.3390/antiox10060891
Chiriaco M, Salfa I, Ursu GM, Cifaldi C, Di Cesare S, Rossi P, Di Matteo G, Finocchi A. Immunological Aspects of X-Linked Chronic Granulomatous Disease Female Carriers. Antioxidants. 2021; 10(6):891. https://doi.org/10.3390/antiox10060891
Chicago/Turabian StyleChiriaco, Maria, Irene Salfa, Giorgiana Madalina Ursu, Cristina Cifaldi, Silvia Di Cesare, Paolo Rossi, Gigliola Di Matteo, and Andrea Finocchi. 2021. "Immunological Aspects of X-Linked Chronic Granulomatous Disease Female Carriers" Antioxidants 10, no. 6: 891. https://doi.org/10.3390/antiox10060891
APA StyleChiriaco, M., Salfa, I., Ursu, G. M., Cifaldi, C., Di Cesare, S., Rossi, P., Di Matteo, G., & Finocchi, A. (2021). Immunological Aspects of X-Linked Chronic Granulomatous Disease Female Carriers. Antioxidants, 10(6), 891. https://doi.org/10.3390/antiox10060891