
DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration




Abstract
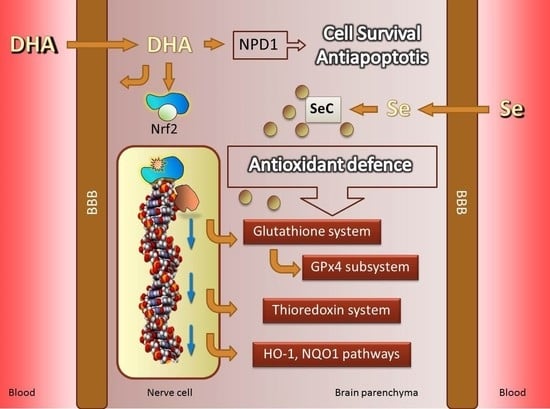
:
1. Introduction
2. DHA Is a Pleiotropic Molecule in Nerve Cells
3. The Hostile Environment of Brain Parenchyma for LCPUFAs
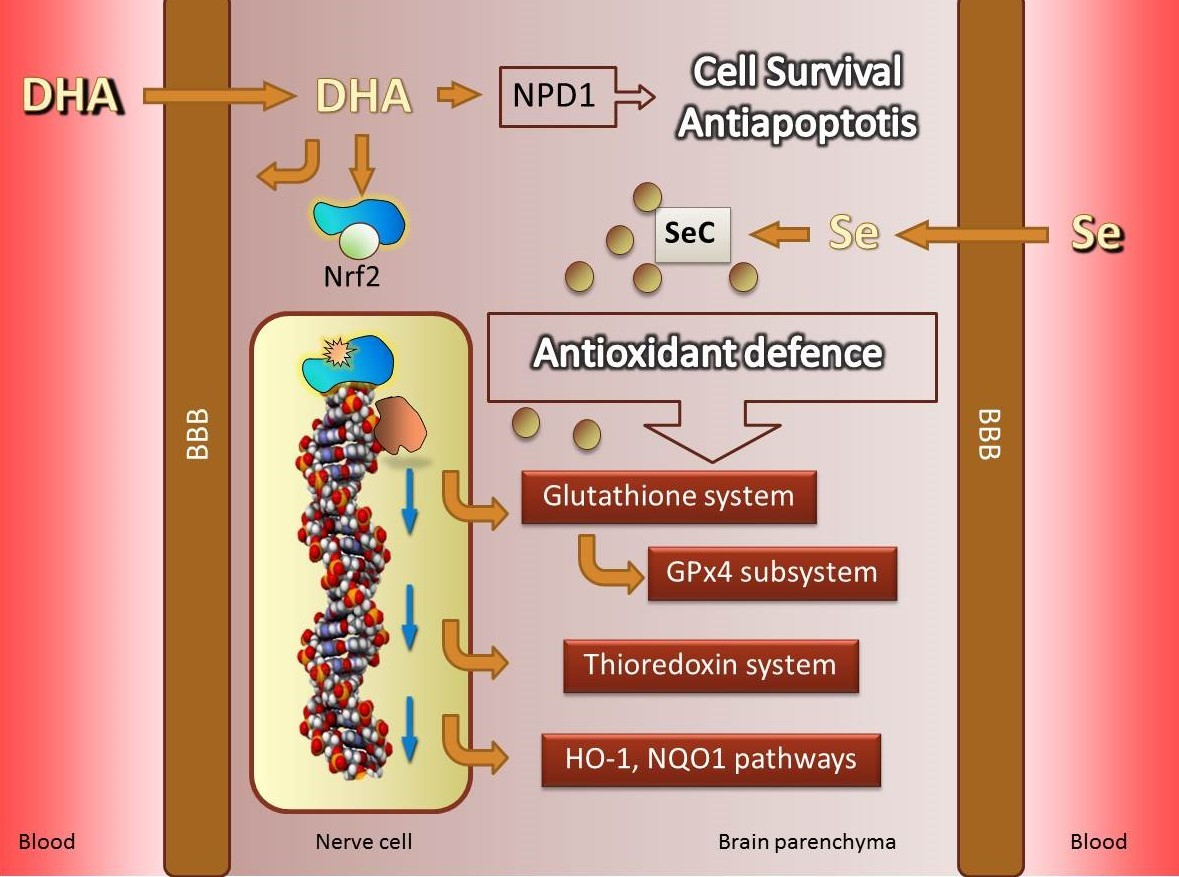
4. Fighting Against Lipid Peroxidation in the Brain
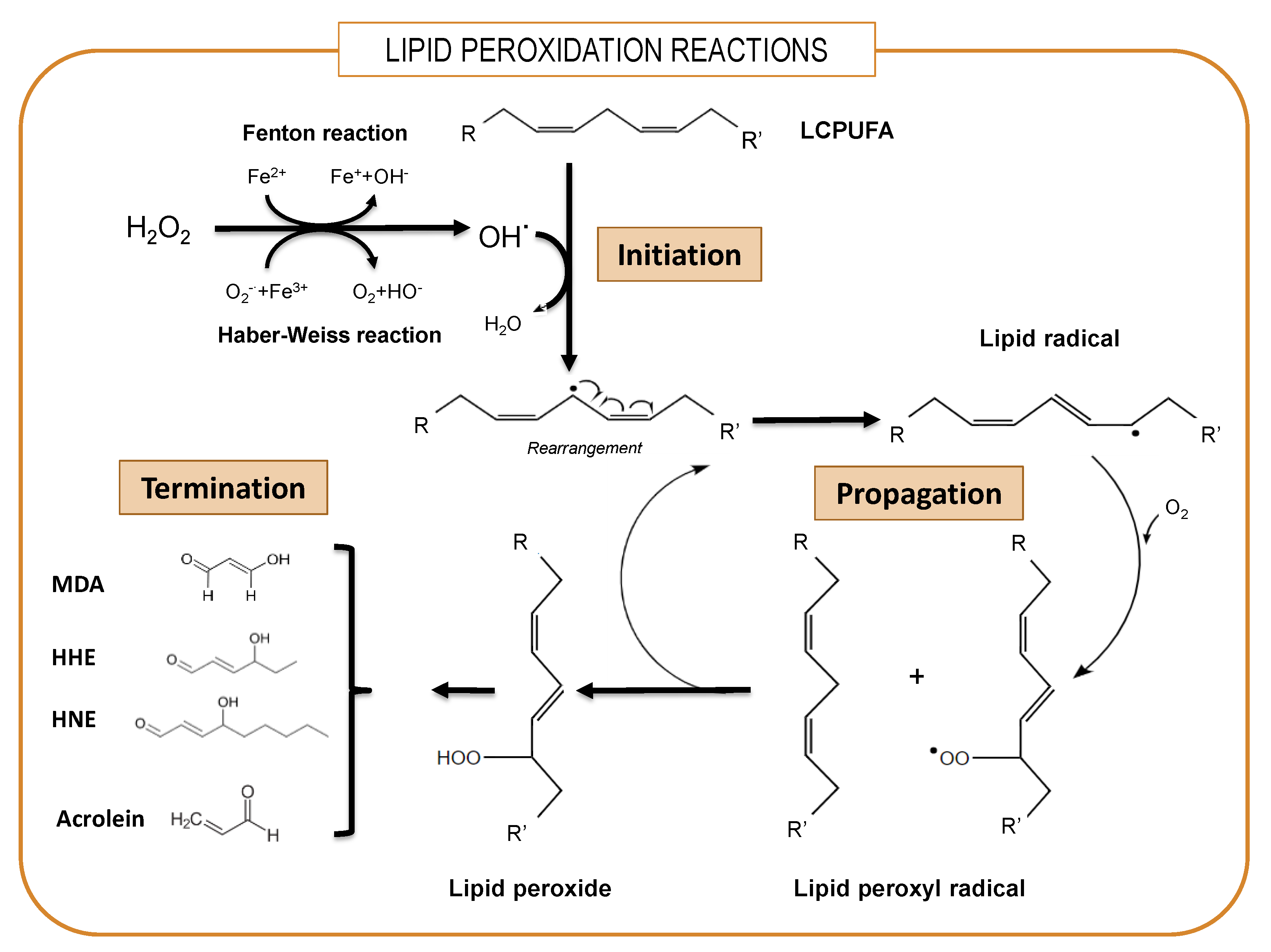
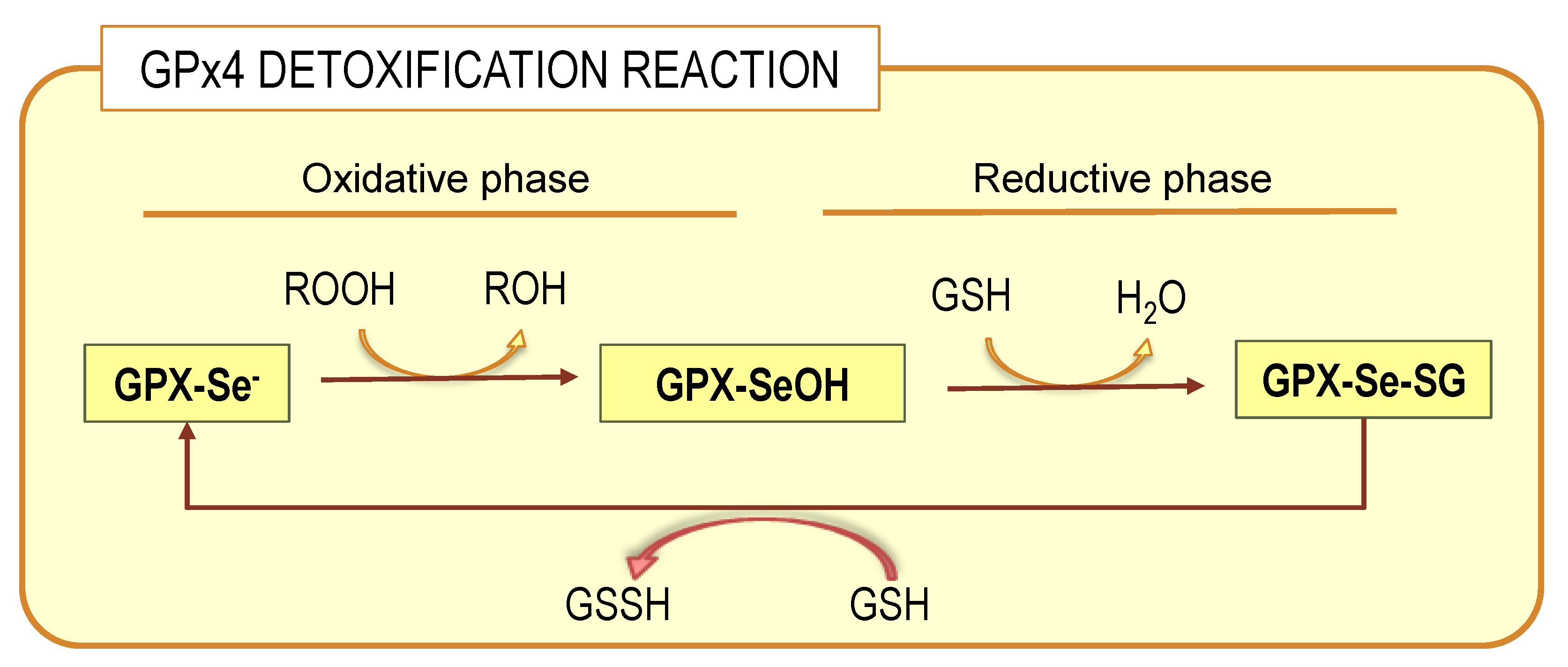
5. The Central Role of GPx4 in the Protection of Membrane LCPUFAs from Peroxidation
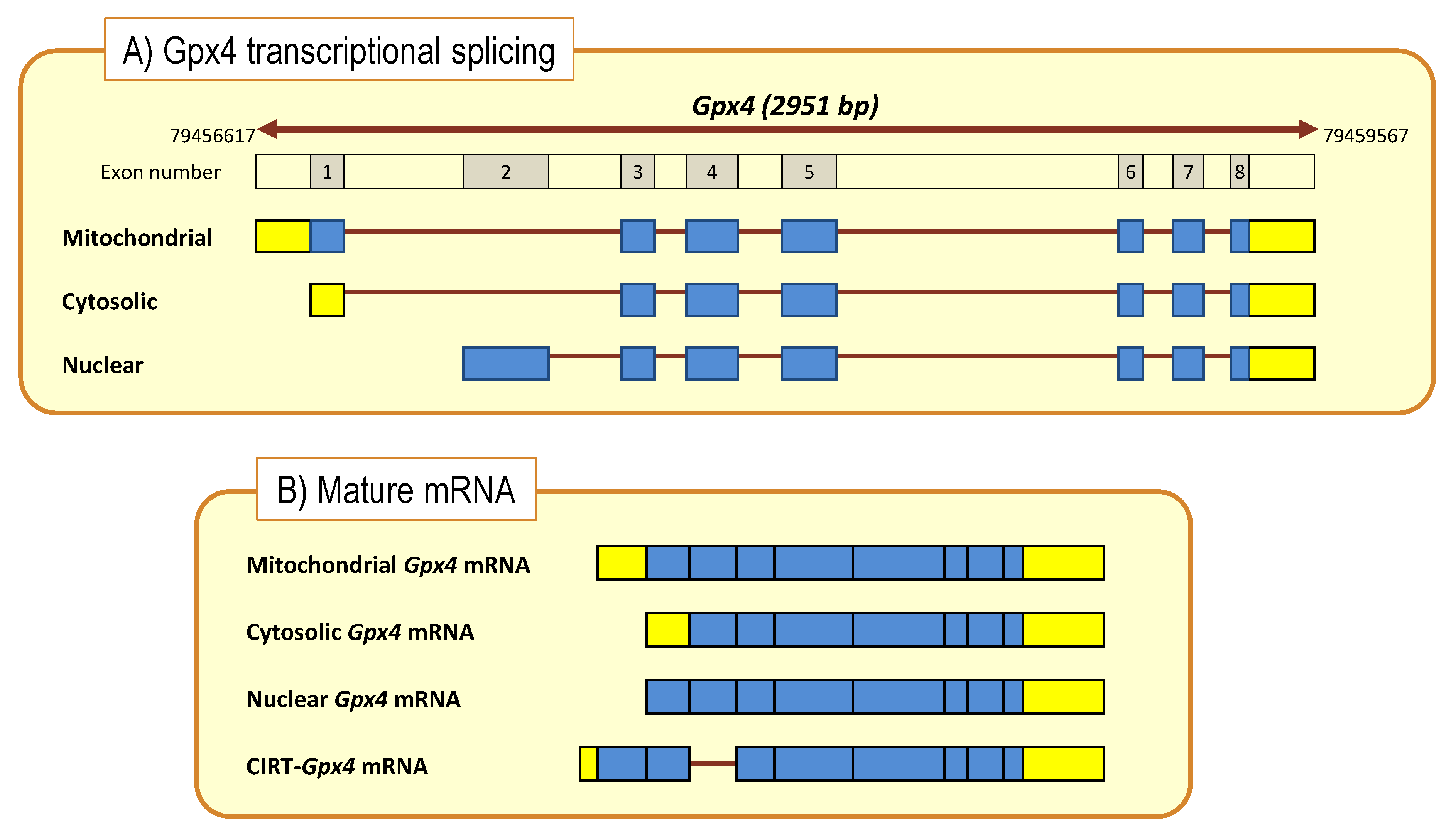
6. The Elaborated Transcriptional Regulation of the Gpx4 Gene by DHA
7. Transcriptional Regulation of Brain Antioxidant Defense by DHA: Beyond GPx4
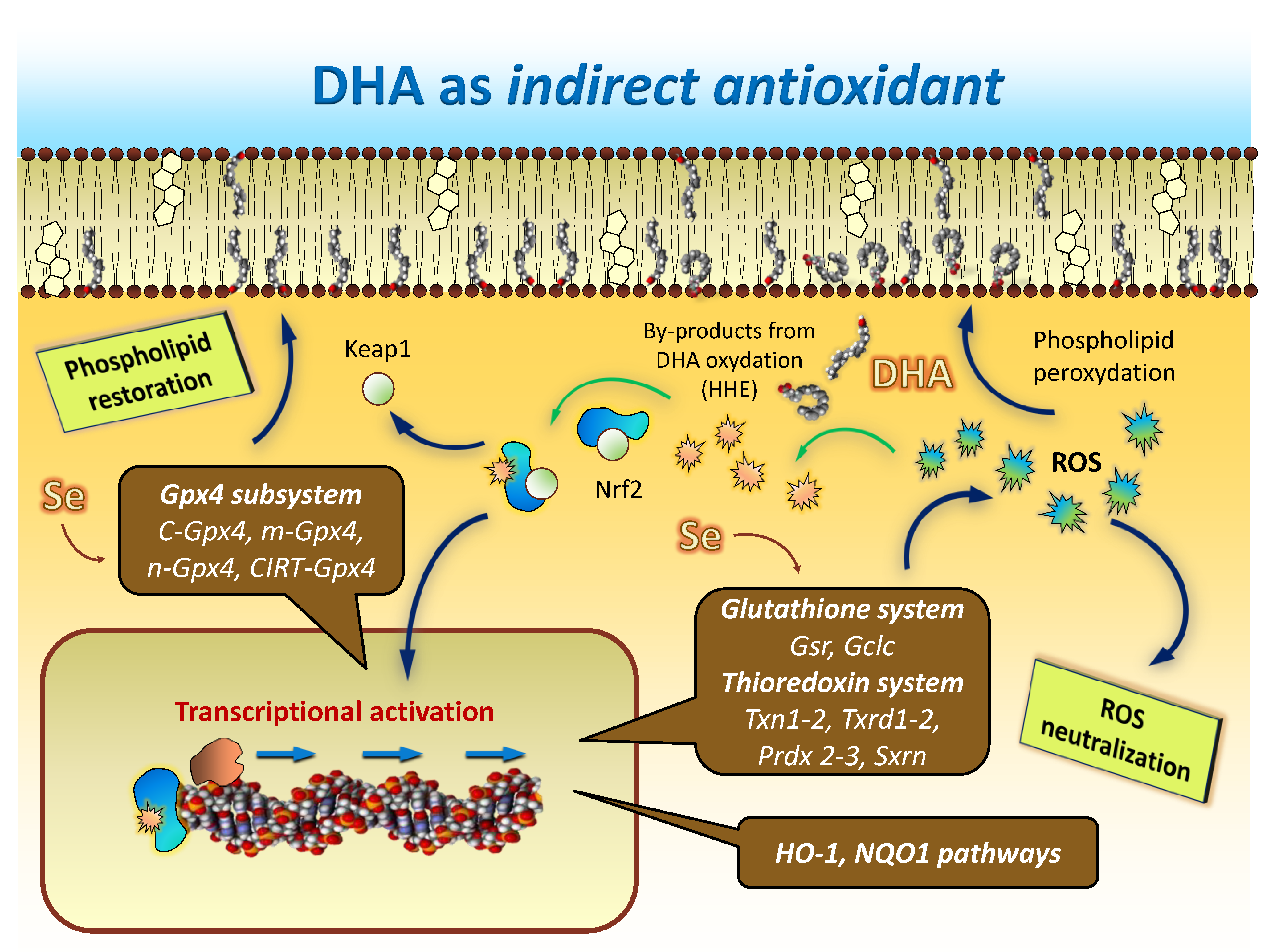
8. DHA: The Ultimate Indirect Antioxidant
9. Selenium Is an Absolute Requirement for Selenoprotein Biosynthesis
10. DHA and GPx4 in Aging and AD Brains
11. Selenium and GPx4: The Fundamental Association
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 12S-HpETE | 12(S)-hydroperoxyeicosatetraenoic |
| 3NT | 3-nitrotyrosine |
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| AA | Arachidonic acid |
| AIF | Apoptosis-inducing factor |
| APP | Amyloid precursor protein |
| ARE | Antioxidant response elements |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| c-GPx4 | Cytosolic glutathione peroxidase 4 |
| CIRTs | Cytoplasmic intron-sequence-retaining transcripts |
| CSF | Cerebrospinal fluid |
| DHA | Docosahexaenoic acid |
| EFsec | Elongation factor |
| EPA | Eicosapentaenoic acid |
| F2-IsoPs | Isoprostane F2 |
| GCLC | Glutamate-cysteine ligase |
| GLRX | Glutaredoxin |
| GPx | Glutathione peroxidase |
| GSR | Glutathione-S-reductase |
| GST | Glutathione-S-transferase |
| HHE | 4-hydroxy-2-trans-hexenal |
| HNE | 4-hydroxy-2-trans-nonenal |
| HO-1 | Heme oxygenase 1 |
| Keap1 | Kelch-like ECH-associated protein |
| L• | Lipid radicals |
| LCPUFAs | Long-chain polyunsaturated fatty acids |
| LOO• | Lipoperoxyl radicals |
| LOOH | Lipoperoxide |
| MCI | Mild cognitive impairment |
| MDA | Malondialdehyde |
| m-GPx4 | Mithocondrial glutathione peroxidase 4 |
| n-GPx4 | Nuclear glutathione peroxidase 4 |
| NPD1 | Neuroprotectin D1 |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid-related factor 2 |
| O2− | Superoxide anion |
| OH• | Hydroxyl radical |
| ONOO− | Peroxynitrite |
| PE | Phosphatidylethanolamine |
| Prdx2 | 2-Cys type peroxiredoxin gene |
| PRDX2 | Cytosolic peroxiredoxin |
| PRDX3 | Mitochondrial peroxiredoxin |
| PS1 | Presenilin 1 |
| PUFAs | Polyunsaturated fatty acids |
| RCS | Reactive carbonyl species |
| RNP | Ribonucleoprotein |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RSL3 | Ras-selective lethal small molecule 3 |
| SBP2 | RNA binding protein 2 |
| SeC | Selenocysteine |
| SelenoP | Selenoproteins |
| SeMet | Selenomethionine |
| SRXN | Sulfiredoxin |
| TXN | Thioredoxin |
| TXNIP | Thioredoxin interacting protein |
| TXNRD | Thioredoxin reductase |
| UOS | Uncontrolled oxidative stress |
References
- Li, Q.; Bozek, K.; Xu, C.; Guo, Y.; Sun, J.; Pääbo, S.; Sherwood, C.C.; Hof, P.R.; Ely, J.J.; Li, Y.; et al. Changes in Lipidome Composition during Brain Development in Humans, Chimpanzees, and Macaque Monkeys. Mol. Biol. Evol. 2017, 34, 1155–1166. [Google Scholar] [CrossRef]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [CrossRef]
- Alessandri, J.-M.; Guesnet, P.; Vancassel, S.; Astorg, P.; Denis, I.; Langelier, B.; Aïd, S.; Poumès-Ballihaut, C.; Champeil-Potokar, G.; Lavialle, M. Polyunsaturated fatty acids in the central nervous system: Evolution of concepts and nutritional implications throughout life. Reprod. Nutr. Dev. 2004, 44, 509–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W.; Wassall, S.R. Docosahexaenoic acid: Membrane properties of a unique fatty acid. Chem. Phys. Lipids 2003, 126, 1–27. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef]
- Díaz, M.; Marín, R. Brain Polyunsaturated Lipids and Neurodegenerative Diseases. In Nutraceuticals and Functional Foods: Natural Remedy; Nova Sciencie: Hauppauge, NY, USA, 2013; pp. 387–412. [Google Scholar]
- Cornelius, F.; Habeck, M.; Kanai, R.; Toyoshima, C.; Karlish, S.J.D. General and specific lipid-protein interactions in Na,K-ATPase. Biochim. Biophys. Acta 2015, 1848, 1729–1743. [Google Scholar] [CrossRef] [Green Version]
- Heberle, F.A.; Feigenson, G.W. Phase separation in lipid membranes. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Foster, L.J.; Chan, Q.W.T. Lipid raft proteomics: More than just detergent-resistant membranes. Subcell. Biochem. 2007, 43, 35–47. [Google Scholar] [CrossRef]
- Suzuki, T.; Zhang, J.; Miyazawa, S.; Liu, Q.; Farzan, M.R.; Yao, W.-D. Association of membrane rafts and postsynaptic density: Proteomics, biochemical, and ultrastructural analyses. J. Neurochem. 2011, 119, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Tsui-Pierchala, B.A.; Encinas, M.; Milbrandt, J.; Johnson, E.M.J. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002, 25, 412–417. [Google Scholar] [CrossRef]
- Shaikh, S.R.; Cherezov, V.; Caffrey, M.; Stillwell, W.; Wassall, S.R. Interaction of cholesterol with a docosahexaenoic acid-containing phosphatidylethanolamine: Trigger for microdomain/raft formation? Biochemistry 2003, 42, 12028–12037. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [Green Version]
- Marin, R. Signalosomes in the brain: Relevance in the development of certain neuropathologies such as Alzheimer’s disease. Front. Physiol. 2011, 2, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon, F.; Kim, H.-Y. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004, 90, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.-S.; Jun, S.B.; Lovinger, D.; Kim, H.-Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef] [Green Version]
- Dagai, L.; Peri-Naor, R.; Birk, R.Z. Docosahexaenoic acid significantly stimulates immediate early response genes and neurite outgrowth. Neurochem. Res. 2009, 34, 867–875. [Google Scholar] [CrossRef]
- Innis, S.M. Dietary (n-3) fatty acids and brain development. J. Nutr. 2007, 137, 855–859. [Google Scholar] [CrossRef] [Green Version]
- Katakura, M.; Hashimoto, M.; Shahdat, H.M.; Gamoh, S.; Okui, T.; Matsuzaki, K.; Shido, O. Docosahexaenoic acid promotes neuronal differentiation by regulating basic helix-loop-helix transcription factors and cell cycle in neural stem cells. Neuroscience 2009, 160, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Asp. Med. 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Qu, X.; Cui, L.; Wang, J.; Kang, J.X. Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc. Natl. Acad. Sci. USA 2009, 106, 11370–11375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriguchi, T.; Harauma, A.; Salem, N.J. Plasticity of mouse brain docosahexaenoic acid: Modulation by diet and age. Lipids 2013, 48, 343–355. [Google Scholar] [CrossRef]
- Calandria, J.M.; Sharp, M.W.; Bazan, N.G. The Docosanoid Neuroprotectin D1 Induces TH-Positive Neuronal Survival in a Cellular Model of Parkinson’s Disease. Cell. Mol. Neurobiol. 2015, 35, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Calon, F. Omega-3 polyunsaturated fatty acids in Alzheimer’s disease: Key questions and partial answers. Curr. Alzheimer Res. 2011, 8, 470–478. [Google Scholar] [CrossRef]
- Fabelo, N.; Martín, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, M.; Edlund, C.; Alafuzoff, I.; Kristensson, K.; Dallner, G. Lipid composition in different regions of the brain in Alzheimer’s disease/senile dementia of Alzheimer’s type. J. Neurochem. 1992, 59, 1646–1653. [Google Scholar] [CrossRef]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 1991, 26, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.-W.; Greenstein, D.; Rapoport, S.I.; Rao, J.S. Brain lipid concentrations in bipolar disorder. J. Psychiatr. Res. 2010, 44, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.-Y.; Huang, S.-Y.; Su, K.-P. A meta-analytic review of polyunsaturated fatty acid compositions in patients with depression. Biol. Psychiatry 2010, 68, 140–147. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K. Long-chain omega-3 fatty acid deficiency in mood disorders: Rationale for treatment and prevention. Curr. Drug Discov. Technol. 2013, 10, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.; Khoutorova, L.; Atkins, K.D.; Bazan, N.G. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke 2009, 40, 3121–3126. [Google Scholar] [CrossRef]
- Ren, C.; Sy, C.; Gao, J.; Ding, Y.; Ji, X. Animal Stroke Model: Ischemia-Reperfusion and Intracerebral Hemorrhage. Methods Mol. Biol. 2016, 1462, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Belayev, L.; Khoutorova, L.; Obenaus, A.; Bazan, N.G. Docosahexaenoic acid confers enduring neuroprotection in experimental stroke. J. Neurol. Sci. 2014, 338, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J. Neurotrauma 2004, 21, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Bazan, N.G.; Musto, A.E.; Knott, E.J. Endogenous signaling by omega-3 docosahexaenoic acid-derived mediators sustains homeostatic synaptic and circuitry integrity. Mol. Neurobiol. 2011, 44, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Bazan, N.G. The docosanoid neuroprotectin D1 induces homeostatic regulation of neuroinflammation and cell survival. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Asatryan, A.; Bazan, N.G. Molecular mechanisms of signaling via the docosanoid neuroprotectin D1 for cellular homeostasis and neuroprotection. J. Biol. Chem. 2017, 292, 12390–12397. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.K.; Chawla, A.; Loayza, M.S.; Bazan, N.G. Docosanoids are multifunctional regulators of neural cell integrity and fate: Significance in aging and disease. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casañas-Sánchez, V.; Pérez, J.A.; Fabelo, N.; Herrera-Herrera, A.V.; Fernández, C.; Marín, R.; González-Montelongo, M.C.; Díaz, M. Addition of docosahexaenoic acid, but not arachidonic acid, activates glutathione and thioredoxin antioxidant systems in murine hippocampal HT22 cells: Potential implications in neuroprotection. J. Neurochem. 2014, 131, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Casañas-Sánchez, V.; Pérez, J.A.; Fabelo, N.; Quinto-Alemany, D.; Díaz, M.L. Docosahexaenoic (DHA) modulates phospholipid-hydroperoxide glutathione peroxidase (Gpx4) gene expression to ensure self-protection from oxidative damage in hippocampal cells. Front. Physiol. 2015, 6, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalá, A.; Díaz, M. Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes; Frontiers Media SA: Lausanne, Switzerland, 2017; ISBN 9782889450824. [Google Scholar]
- Díaz, M.; Casañas-Sánchez, V.; Marín, R.; Pérez, J.A. Fighting against Lipid Peroxidation in the Brain: The Unique Story of Docosahexaenoic Acid. In Lipid Peroxidation: Inhibition, Effects and Mechanisms; Catalá, A., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2017; pp. 15–26. [Google Scholar]
- Bazan, N.G. Docosanoids and elovanoids from omega-3 fatty acids are pro-homeostatic modulators of inflammatory responses, cell damage and neuroprotection. Mol. Asp. Med. 2018, 64, 18–33. [Google Scholar] [CrossRef]
- Belayev, L.; Khoutorova, L.; Atkins, K.D.; Eady, T.N.; Hong, S.; Lu, Y.; Obenaus, A.; Bazan, N.G. Docosahexaenoic Acid therapy of experimental ischemic stroke. Transl. Stroke Res. 2011, 2, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayurasakorn, K.; Williams, J.J.; Ten, V.S.; Deckelbaum, R.J. Docosahexaenoic acid: Brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Pelaez, R.; Lukiw, W.J.; Bazan, N.G. Omega-3 essential fatty acids modulate initiation and progression of neurodegenerative disease. Mol. Neurobiol. 2010, 41, 367–374. [Google Scholar] [CrossRef]
- Yamagata, K. Dietary docosahexaenoic acid inhibits neurodegeneration and prevents stroke. J. Neurosci. Res. 2021, 99, 561–572. [Google Scholar] [CrossRef]
- Kim, H.Y.; Akbar, M.; Kim, K.Y. Inhibition of neuronal apoptosis by polyunsaturated fatty acids. J. Mol. Neurosci. 2001, 16, 223–284. [Google Scholar] [CrossRef]
- Díaz, M.; Fabelo, N.; Martín, V.; Ferrer, I.; Gómez, T.; Marín, R. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef]
- Yang, X.; Askarova, S.; Lee, J.C.-M. Membrane biophysics and mechanics in Alzheimer’s disease. Mol. Neurobiol. 2010, 41, 138–148. [Google Scholar] [CrossRef]
- Lauritzen, L.; Brambilla, P.; Mazzocchi, A.; Harsløf, L.B.S.; Ciappolino, V.; Agostoni, C. DHA Effects in Brain Development and Function. Nutrients 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouinard-Watkins, R.; Chen, C.T.; Metherel, A.H.; Lacombe, R.J.S.; Thies, F.; Masoodi, M.; Bazinet, R.P. Phospholipid class-specific brain enrichment in response to lysophosphatidylcholine docosahexaenoic acid infusion. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Fabelo, N.; Casañas-Sánchez, V.; Marin, R.; Gómez, T.; Quinto-Alemany, D.; Pérez, J.A. Hippocampal Lipid Homeostasis in APP/PS1 Mice is Modulated by a Complex Interplay Between Dietary DHA and Estrogens: Relevance for Alzheimer’s Disease. J. Alzheimers Dis. 2016, 49, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Kitson, A.P.; Stark, K.D.; Duncan, R.E. Enzymes in brain phospholipid docosahexaenoic acid accretion: A PL-ethora of potential PL-ayers. Prostaglandins Leukot. Essent. Fatty Acids 2012, 87, 1–10. [Google Scholar] [CrossRef]
- Huang, T.L. Omega-3 fatty acids, cognitive decline, and Alzheimer’s disease: A critical review and evaluation of the literature. J. Alzheimers Dis. 2010, 21, 673–690. [Google Scholar] [CrossRef]
- Akerele, O.A.; Cheema, S.K. Maternal diet high in Omega-3 fatty acids upregulate genes involved in neurotrophin signalling in fetal brain during pregnancy in C57BL/6 mice. Neurochem. Int. 2020, 138, 1–28. [Google Scholar] [CrossRef]
- Bang, H.-Y.; Park, S.-A.; Saeidi, S.; Na, H.K.; Surh, Y.-J. Docosahexaenoic Acid Induces Expression of Heme Oxygenase-1 and NAD(P)H:quinone Oxidoreductase through Activation of Nrf2 in Human Mammary Epithelial Cells. Molecules 2017, 22, 969. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Li, M.-Y.; Li, G.; Li, S.-J.; Wen, B.; Lu, Y.; Yu, X. Retinoid X Receptor α Regulates DHA-Dependent Spinogenesis and Functional Synapse Formation In Vivo. Cell Rep. 2020, 31, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Kato, A.; Sango, K.; Himeno, T.; Kondo, M.; Kato, Y.; Kamiya, H.; Nakamura, J.; Kato, K. Omega-3 polyunsaturated fatty acids exert anti-oxidant effects through the nuclear factor (erythroid-derived 2)-related factor 2 pathway in immortalized mouse Schwann cells. J. Diabetes Investig. 2019, 10, 602–612. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Walder, K.; Puri, B.K.; Berk, M.; Maes, M. The Deleterious Effects of Oxidative and Nitrosative Stress on Palmitoylation, Membrane Lipid Rafts and Lipid-Based Cellular Signalling: New Drug Targets in Neuroimmune Disorders. Mol. Neurobiol. 2016, 53, 4638–4658. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Salminen, L.E.; Paul, R.H. Oxidative stress and genetic markers of suboptimal antioxidant defense in the aging brain: A theoretical review. Rev. Neurosci. 2014, 25, 805–819. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1–15. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Borchert, A.; Bräuer, A.U.; Kuhn, H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic. Biol. Med. 2007, 43, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Glutathione Peroxidases. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 399–404. ISBN 978-0-12-378631-9. [Google Scholar]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Janowski, R.; Scanu, S.; Niessing, D.; Madl, T. Crystal and solution structural studies of mouse phospholipid hydroperoxide glutathione peroxidase 4. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Scheerer, P.; Borchert, A.; Krauss, N.; Wessner, H.; Gerth, C.; Höhne, W.; Kuhn, H. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry 2007, 46, 9041–9049. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Ufer, C.; Kühn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Buckley, P.T.; Khaladkar, M.; Kim, J.; Eberwine, J. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip. Rev. RNA 2014, 5, 223–230. [Google Scholar] [CrossRef]
- Khaladkar, M.; Buckley, P.T.; Lee, M.T.; Francis, C.; Eghbal, M.M.; Chuong, T.; Suresh, S.; Kuhn, B.; Eberwine, J.; Kim, J. Subcellular RNA sequencing reveals broad presence of cytoplasmic intron-sequence retaining transcripts in mouse and rat neurons. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bell, T.J.; Miyashiro, K.Y.; Sul, J.-Y.; McCullough, R.; Buckley, P.T.; Jochems, J.; Meaney, D.F.; Haydon, P.; Cantor, C.; Parsons, T.D.; et al. Cytoplasmic BK(Ca) channel intron-containing mRNAs contribute to the intrinsic excitability of hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 1901–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.J.; Miyashiro, K.Y.; Sul, J.-Y.; Buckley, P.T.; Lee, M.T.; McCullough, R.; Jochems, J.; Kim, J.; Cantor, C.R.; Parsons, T.D.; et al. Intron retention facilitates splice variant diversity in calcium-activated big potassium channel populations. Proc. Natl. Acad. Sci. USA 2010, 107, 21152–21157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Liu, J.R.; Kyle, J.W.; Hanck, D.A.; Agnew, W.S. A profile of alternative RNA splicing and transcript variation of CACNA1H, a human T-channel gene candidate for idiopathic generalized epilepsies. Hum. Mol. Genet. 2006, 15, 1497–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharangdhar, T.; Sugimoto, Y.; Heraud-Farlow, J.; Fernández-Moya, S.M.; Ehses, J.; Ruiz de Los Mozos, I.; Ule, J.; Kiebler, M.A. A retained intron in the 3’-UTR of Calm3 mRNA mediates its Staufen2- and activity-dependent localization to neuronal dendrites. EMBO Rep. 2017, 18, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.; Kiebler, M.A. Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J. 2011, 30, 3540–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, S.; Kreienkamp, H.-J. Dendritic mRNA targeting and translation. Adv. Exp. Med. Biol. 2012, 970, 285–305. [Google Scholar] [CrossRef]
- Jutzi, D.; Akinyi, M.V.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.-D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Björnstedt, M.; Hamberg, M.; Kumar, S.; Xue, J.; Holmgren, A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J. Biol. Chem. 1995, 270, 11761–11764. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Karplus, P.A.; Poole, L.B. Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. FEBS J. 2009, 276, 2469–2477. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.; Bae, S.H.; Toledano, M.B.; Rhee, S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 2012, 53, 447–456. [Google Scholar] [CrossRef]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.-K.; Hong, J.T. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Radyuk, S.N.; Orr, W.C. The Multifaceted Impact of Peroxiredoxins on Aging and Disease. Antioxid. Redox Signal. 2018, 29, 1293–1311. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, S.; Mao, L.; Leak, R.K.; Shi, Y.; Zhang, W.; Hu, X.; Sun, B.; Cao, G.; Gao, Y.; et al. Omega-3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J. Neurosci. 2014, 34, 1903–1915. [Google Scholar] [CrossRef]
- Zhu, W.; Ding, Y.; Kong, W.; Li, T.; Chen, H. Docosahexaenoic Acid (DHA) Provides Neuroprotection in Traumatic Brain Injury Models via Activating Nrf2-ARE Signaling. Inflammation 2018, 41, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tanji, K.; Wakabayashi, K.; Matsuura, S.; Itoh, K. Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol. Int. 2015, 65, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52 Suppl 1, S128–S138. [Google Scholar] [CrossRef]
- Valentová, K. Cytoprotective Activity of Natural and Synthetic Antioxidants. Antioxidants 2020, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Kozarski, M.; Klaus, A.; Jakovljevic, D.; Todorovic, N.; Vunduk, J.; Petrović, P.; Niksic, M.; Vrvic, M.M.; van Griensven, L. Antioxidants of Edible Mushrooms. Molecules 2015, 20, 19489–19525. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.N.; Mishra, D.; Singh, P.; Vamanu, E.; Singh, M.P. Therapeutic applications of mushrooms and their biomolecules along with a glimpse of in silico approach in neurodegenerative diseases. Biomed. Pharmacother. 2021, 137, 1–14. [Google Scholar] [CrossRef]
- Gladyshev, M.I.; Sushchik, N.N. Long-chain Omega-3 Polyunsaturated Fatty Acids in Natural Ecosystems and the Human Diet: Assumptions and Challenges. Biomolecules 2019, 9, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tocher, D.R.; Betancor, M.B.; Sprague, M.; Olsen, R.E.; Napier, J.A. Omega-3 Long-Chain Polyunsaturated Fatty Acids, EPA and DHA: Bridging the Gap between Supply and Demand. Nutrients 2019, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Zgórzyńska, E.; Dziedzic, B.; Gorzkiewicz, A.; Stulczewski, D.; Bielawska, K.; Su, K.-P.; Walczewska, A. Omega-3 polyunsaturated fatty acids improve the antioxidative defense in rat astrocytes via an Nrf2-dependent mechanism. Pharmacol. Rep. 2017, 69, 935–942. [Google Scholar] [CrossRef]
- Van Kuijk, F.J.; Holte, L.L.; Dratz, E.A. 4-Hydroxyhexenal: A lipid peroxidation product derived from oxidized docosahexaenoic acid. Biochim. Biophys. Acta 1990, 1043, 116–118. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Tyurin, V.A.; Anthonymuthu, T.; Amoscato, A.A.; Sparvero, L.J.; Nesterova, A.M.; Baynard, M.L.; Sun, W.; He, R.; Khaitovich, P.; et al. Redox lipidomics technology: Looking for a needle in a haystack. Chem. Phys. Lipids 2019, 221, 93–107. [Google Scholar] [CrossRef]
- Ishikado, A.; Morino, K.; Nishio, Y.; Nakagawa, F.; Mukose, A.; Sono, Y.; Yoshioka, N.; Kondo, K.; Sekine, O.; Yoshizaki, T.; et al. 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, C.; Yang, L.; Yoshizaki, T.; Nakagawa, F.; Ishikado, A.; Kondo, M.; Morino, K.; Sekine, O.; Ugi, S.; Nishio, Y.; et al. Omega-3 polyunsaturated fatty acid has an anti-oxidant effect via the Nrf-2/HO-1 pathway in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2013, 430, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Metallomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, R.; Uyehara-Lock, J.H.; Bellinger, F.P. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Sies, H. Selenium homeostasis and antioxidant selenoproteins in brain: Implications for disorders in the central nervous system. Arch. Biochem. Biophys. 2013, 536, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.J.; Worland, P.J.; Davis, J.N.; Stadtman, T.C.; Hatfield, D.L. Identification of a selenocysteyl-tRNA(Ser) in mammalian cells that recognizes the nonsense codon, UGA. J. Biol. Chem. 1989, 264, 9724–9727. [Google Scholar] [CrossRef]
- Allmang, C.; Krol, A. Selenoprotein Biosynthesis BT-Selenoproteins and Mimics; Liu, J., Luo, G., Mu, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 107–124. ISBN 978-3-642-22236-8. [Google Scholar]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef]
- Rose, M.R. Adaptation, aging, and genomic information. Aging (Albany. NY). 2009, 1, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. Organo Hosp. Enfermedades Nutr. 2016, 68, 7–16. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barja, G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124, 1–14. [Google Scholar] [CrossRef]
- Cabré, R.; Naudí, A.; Dominguez-Gonzalez, M.; Jové, M.; Ayala, V.; Mota-Martorell, N.; Pradas, I.; Nogueras, L.; Rué, M.; Portero-Otín, M.; et al. Lipid Profile in Human Frontal Cortex Is Sustained Throughout Healthy Adult Life Span to Decay at Advanced Ages. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxidative Med. Cell. Longev. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Peskind, E.R.; Quinn, J.F.; Wilson, A.M.; Montine, K.S.; Galasko, D. Increased cerebrospinal fluid F2-isoprostanes are associated with aging and latent Alzheimer’s disease as identified by biomarkers. Neuromol. Med. 2011, 13, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Praticò, D.; Clark, C.M.; Lee, V.M.; Trojanowski, J.Q.; Rokach, J.; FitzGerald, G.A. Increased 8,12-iso-iPF2alpha-VI in Alzheimer’s disease: Correlation of a noninvasive index of lipid peroxidation with disease severity. Ann. Neurol. 2000, 48, 809–812. [Google Scholar] [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Mattson, M.P.; Markesbery, W.R. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol. Aging 1997, 18, 457–461. [Google Scholar] [CrossRef]
- Colin, J.; Gregory-Pauron, L.; Lanhers, M.-C.; Claudepierre, T.; Corbier, C.; Yen, F.T.; Malaplate-Armand, C.; Oster, T. Membrane raft domains and remodeling in aging brain. Biochimie 2016, 130, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Fabelo, N.; Ferrer, I.; Marín, R. “Lipid raft aging” in the human frontal cortex during nonpathological aging: Gender influences and potential implications in Alzheimer’s disease. Neurobiol. Aging 2018, 67, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabelo, N.; Martín, V.; Marín, R.; Santpere, G.; Aso, E.; Ferrer, I.; Díaz, M. Evidence for premature lipid raft aging in APP/PS1 double-transgenic mice, a model of familial Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 868–881. [Google Scholar] [CrossRef] [Green Version]
- Mesa-Herrera, F.; Taoro-González, L.; Valdés-Baizabal, C.; Diaz, M.; Marín, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef] [Green Version]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 1–28. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Cipolla, M.; Chiang, J.; Arakaki, X.; Harrington, M.G. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS ONE 2014, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.R.; Lovell, M.A.; Yatin, M.; Dhillon, H.; Markesbery, W.R. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 1998, 23, 81–88. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ong, W.-Y.; Horrocks, L.A. Biochemical aspects of neurodegeneration in human brain: Involvement of neural membrane phospholipids and phospholipases A2. Neurochem. Res. 2004, 29, 1961–1977. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Conquer, J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod. Nutr. Dev. 2005, 45, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Bungart, B.L.; Yang, X.; Zhumadilov, Z.; Lee, J.C.-M.; Askarova, S. Role of membrane biophysics in Alzheimer’s-related cell pathways. Front. Neurosci. 2015, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, M.L.; Fabelo, N.; Marín, R. Genotype-induced changes in biophysical properties of frontal cortex lipid raft from APP/PS1 transgenic mice. Front. Physiol. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, G.; Díaz, M.; Torres, N. V Lipid Raft Size and Lipid Mobility in Non-raft Domains Increase during Aging and Are Exacerbated in APP/PS1 Mice Model of Alzheimer’s Disease. Predictions from an Agent-Based Mathematical Model. Front. Physiol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, R.; Kawai, T. Cerebrospinal fluid oxidative stress markers in Alzheimer’s disease. Neurol. Clin. Neurosci. 2020, 8, 232–240. [Google Scholar] [CrossRef]
- Gmitterová, K.; Heinemann, U.; Gawinecka, J.; Varges, D.; Ciesielczyk, B.; Valkovic, P.; Benetin, J.; Zerr, I. 8-OHdG in cerebrospinal fluid as a marker of oxidative stress in various neurodegenerative diseases. Neurodegener. Dis. 2009, 6, 263–269. [Google Scholar] [CrossRef]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef]
- Calabrese, V.; Sultana, R.; Scapagnini, G.; Guagliano, E.; Sapienza, M.; Bella, R.; Kanski, J.; Pennisi, G.; Mancuso, C.; Stella, A.M.G.; et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid. Redox Signal. 2006, 8, 1975–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannan, T.S.; Maker, H.S.; Weiss, C.; Cohen, G. Regional distribution of glutathione peroxidase in the adult rat brain. J. Neurochem. 1980, 35, 1013–1014. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- Cieślik, M.; Czapski, G.A.; Wójtowicz, S.; Wieczorek, I.; Wencel, P.L.; Strosznajder, R.P.; Jaber, V.; Lukiw, W.J.; Strosznajder, J.B. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid β Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1374–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, J.H.T.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Gu, M.; Van Remmen, H.; Strong, R.; Roberts, J.L.; Richardson, A. Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J. Neurosci. Res. 2006, 84, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Na, R.; Gu, M.; Richardson, A.; Ran, Q. Lipid peroxidation up-regulates BACE1 expression in vivo: A possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem. 2008, 107, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Lei, J.; Chen, Z.; Song, S.; Sheng, C.; Song, S.; Zhu, J. Insight Into the Role of Ferroptosis in Non-neoplastic Neurological Diseases. Front. Cell. Neurosci. 2020, 14, 231. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Ran, Q.; Liang, H. The Use of Gpx4 Knockout Mice and Transgenic Mice to Study the Roles of Lipid Peroxidation in Diseases and Aging BT-Studies on Experimental Models; Basu, S., Wiklund, L., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 265–278. ISBN 978-1-60761-956-7. [Google Scholar]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zheng, S.G. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front. Immunol. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Vučković, A.-M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; Song, W.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Haratake, M.; Yoshida, S.; Mandai, M.; Fuchigami, T.; Nakayama, M. Elevated amyloid-β plaque deposition in dietary selenium-deficient Tg2576 transgenic mice. Metallomics 2013, 5, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Solovyev, N.D. Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Vázquez-Román, B.; La Cruz, V.P.-D.; González-Cortés, C.; Trejo-Solís, M.C.; Galván-Arzate, S.; Jara-Prado, A.; Guevara-Fonseca, J.; Ali, S.F. Selenium reduces the proapoptotic signaling associated to NF-kappaB pathway and stimulates glutathione peroxidase activity during excitotoxic damage produced by quinolinate in rat corpus striatum. Synapse 2005, 58, 258–266. [Google Scholar] [CrossRef]
- Kühbacher, M.; Bartel, J.; Hoppe, B.; Alber, D.; Bukalis, G.; Bräuer, A.U.; Behne, D.; Kyriakopoulos, A. The brain selenoproteome: Priorities in the hierarchy and different levels of selenium homeostasis in the brain of selenium-deficient rats. J. Neurochem. 2009, 110, 133–142. [Google Scholar] [CrossRef]
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Almeida, A. Anatomical regional differences in selenium levels in the human brain. Biol. Trace Elem. Res. 2015, 163, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Rocourt, C.; Cheng, W.-H. Selenoproteins and the aging brain. Mech. Ageing Dev. 2010, 131, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Hare, D.J.; Macpherson, H. Sex-dependent association between selenium status and cognitive performance in older adults. Eur. J. Nutr. 2021, 60, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Ong, T.P.; Jacob-Filho, W.; Jaluul, O.; d’Avila Freitas, M.I.; Cozzolino, S.M.F. Nutritional status of selenium in Alzheimer’s disease patients. Br. J. Nutr. 2010, 103, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Jin, Y.; Hall, K.S.; Liang, C.; Unverzagt, F.W.; Ji, R.; Murrell, J.R.; Cao, J.; Shen, J.; Ma, F.; et al. Selenium level and cognitive function in rural elderly Chinese. Am. J. Epidemiol. 2007, 165, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, B.R.; Apolinário, D.; da Silva Bandeira, V.; Busse, A.L.; Magaldi, R.M.; Jacob-Filho, W.; Cozzolino, S.M.F. Effects of Brazil nut consumption on selenium status and cognitive performance in older adults with mild cognitive impairment: A randomized controlled pilot trial. Eur. J. Nutr. 2016, 55, 107–116. [Google Scholar] [CrossRef]
- Loef, M.; Schrauzer, G.N.; Walach, H. Selenium and Alzheimer’s disease: A systematic review. J. Alzheimers Dis. 2011, 26, 81–104. [Google Scholar] [CrossRef] [Green Version]
- Ishrat, T.; Parveen, K.; Khan, M.M.; Khuwaja, G.; Khan, M.B.; Yousuf, S.; Ahmad, A.; Shrivastav, P.; Islam, F. Selenium prevents cognitive decline and oxidative damage in rat model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Res. 2009, 1281, 117–127. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xiong, S.; Lyubartseva, G.; Markesbery, W.R. Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic. Biol. Med. 2009, 46, 1527–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornett, C.R.; Markesbery, W.R.; Ehmann, W.D. Imbalances of trace elements related to oxidative damage in Alzheimer’s disease brain. Neurotoxicology 1998, 19, 339–345. [Google Scholar]
- Tamtaji, O.R.; Heidari-Soureshjani, R.; Mirhosseini, N.; Kouchaki, E.; Bahmani, F.; Aghadavod, E.; Tajabadi-Ebrahimi, M.; Asemi, Z. Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: A randomized, double-blind, controlled trial. Clin. Nutr. 2019, 38, 2569–2575. [Google Scholar] [CrossRef]
- Gerhardsson, L.; Blennow, K.; Lundh, T.; Londos, E.; Minthon, L. Concentrations of metals, beta-amyloid and tau-markers in cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Battin, E.E.; Zimmerman, M.T.; Ramoutar, R.R.; Quarles, C.E.; Brumaghim, J.L. Preventing metal-mediated oxidative DNA damage with selenium compounds. Metallomics 2011, 3, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Ramoutar, R.R.; Brumaghim, J.L. Effects of inorganic selenium compounds on oxidative DNA damage. J. Inorg. Biochem. 2007, 101, 1028–1035. [Google Scholar] [CrossRef]
- Khalili, H.; Ahl, R.; Cao, Y.; Paydar, S.; Sjölin, G.; Niakan, A.; Dabiri, G.; Mohseni, S. Early selenium treatment for traumatic brain injury: Does it improve survival and functional outcome? Injury 2017, 48, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Dalla Puppa, L.; Savaskan, N.E.; Bräuer, A.U.; Behne, D.; Kyriakopoulos, A. The role of selenite on microglial migration. Ann. N. Y. Acad. Sci. 2007, 1096, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uğuz, A.C.; Nazıroğlu, M. Effects of selenium on calcium signaling and apoptosis in rat dorsal root ganglion neurons induced by oxidative stress. Neurochem. Res. 2012, 37, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, H.; Wang, Z.; Qiu, S.; Liu, Q.; Ni, J. Selenoprotein P and selenoprotein M block Zn2+ -mediated Aβ42 aggregation and toxicity. Metallomics 2013, 5, 861–870. [Google Scholar] [CrossRef]
- Du, X.; Wang, C.; Liu, Q. Potential Roles of Selenium and Selenoproteins in the Prevention of Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 835–848. [Google Scholar] [CrossRef]
- Yim, S.Y.; Chae, K.R.; Shim, S.B.; Hong, J.T.; Park, J.Y.; Lee, C.Y.; Son, H.J.; Sheen, Y.Y.; Hwang, D.Y. ERK activation induced by selenium treatment significantly downregulates beta/gamma-secretase activity and Tau phosphorylation in the transgenic rat overexpressing human selenoprotein M. Int. J. Mol. Med. 2009, 24, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Godoi, G.L.; de Oliveira Porciúncula, L.; Schulz, J.F.; Kaufmann, F.N.; da Rocha, J.B.; de Souza, D.O.G.; Ghisleni, G.; de Almeida, H.L.J. Selenium compounds prevent amyloid β-peptide neurotoxicity in rat primary hippocampal neurons. Neurochem. Res. 2013, 38, 2359–2363. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, N.M.; Martin, D.; Hutter-Paier, B.; Windisch, M.; Nguyen, T.; Nheu, L.; Sundstrom, L.E.; Costello, A.J.; Hovens, C.M. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2010, 17, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Götz, J.; Ittner, L.M. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Markesbery, W.R.; Shao, C.; Lovell, M.A. Seleno-L-methionine protects against beta-amyloid and iron/hydrogen peroxide-mediated neuron death. Antioxid. Redox Signal. 2007, 9, 457–467. [Google Scholar] [CrossRef]
- Song, G.; Zhang, Z.; Wen, L.; Chen, C.; Shi, Q.; Zhang, Y.; Ni, J.; Liu, Q. Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 85–99. [Google Scholar] [CrossRef]
- Basun, H.; Forssell, L.G.; Wetterberg, L.; Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 1991, 3, 231–258. [Google Scholar]
- Mesa-Herrera, F. Nuevos Abordajes Neuroquímicos del Líquido Cefalorraquídeo Orientados a la Detección Precoz de la Enfermedad de Alzheimer; Universidad de La Laguna: Santa Cruz de Tenerife, Spain, 2020. [Google Scholar]
- Gerhardsson, L.; Lundh, T.; Minthon, L.; Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 25, 508–515. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohé, L.; Sendtner, M.; Köhrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinceti, M.; Chiari, A.; Eichmüller, M.; Rothman, K.J.; Filippini, T.; Malagoli, C.; Weuve, J.; Tondelli, M.; Zamboni, G.; Nichelli, P.F.; et al. A selenium species in cerebrospinal fluid predicts conversion to Alzheimer’s dementia in persons with mild cognitive impairment. Alzheimers Res. Ther. 2017, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Johanson, C.; McMillan, P.; Tavares, R.; Spangenberger, A.; Duncan, J.; Silverberg, G.; Stopa, E. Homeostatic capabilities of the choroid plexus epithelium in Alzheimer’s disease. Cerebrospinal Fluid Res. 2004, 1, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Examples | References |
|---|---|---|
| Bilayer Asymmetry | DHA-enriched PE: Inner leaflet >> Outer leaflet | [3,5,6,8,9,15] |
| Organization of Membrane Domains | Segregation of lipid rafts and non-raft microdomains | [5,6,9,10,15] |
| Biophysical Properties | Membrane fluidity, thermodynamic of lipid–protein interactions | [5,6,10,53,54,55] |
| Nerve Cell Signaling | Neuroprotectin D1/PI3K/Akt pathway | [4,39,41,42] |
| Neurodevelopment | Fetal and Postnatal Brain accretion | [3,21,56] |
| Neuromodulation | Synaptogenesis, LTP, dendritogenesis, neurotransmission | [3,4,18,19,20,21,22,23,24,25] |
| Anti-inflammatory Mediators | Docosanoids, elovanoids | [4,39,40,47] |
| Modulation of Apoptosis | Induction of anti-apoptotic Bcl-2 family of proteins | [4,40,41,47,48,49,51,52] |
| Regulation of Lipid Metabolism | Cholesterol biosynthesis, phospholipid remodeling | [57,58,59] |
| Neuroprotection | Processing of amyloid precursor protein (APP) | [7,26,35,36,37,50,54,60] |
| Modulation of Gene Transcription | Interaction with PPAR/RXR/Nrf2 transcription factors | [22,61,62,63,64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, M.; Mesa-Herrera, F.; Marín, R. DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration. Antioxidants 2021, 10, 907. https://doi.org/10.3390/antiox10060907
Díaz M, Mesa-Herrera F, Marín R. DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration. Antioxidants. 2021; 10(6):907. https://doi.org/10.3390/antiox10060907
Chicago/Turabian StyleDíaz, Mario, Fátima Mesa-Herrera, and Raquel Marín. 2021. "DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration" Antioxidants 10, no. 6: 907. https://doi.org/10.3390/antiox10060907
APA StyleDíaz, M., Mesa-Herrera, F., & Marín, R. (2021). DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration. Antioxidants, 10(6), 907. https://doi.org/10.3390/antiox10060907