
Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation


 , ,
, , 
 and
and 
Abstract
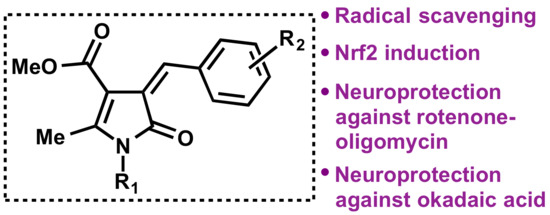
:
1. Introduction
2. Materials and Methods
2.1. Chemistry: General Information
2.2. Synthesis of 2-Pyrrolin-5-one Derivatives 1
2.3. Synthesis of Non-Commercial 4-benzyloxy-3-methoxybenzaldehydes 2
2.3.1. 4-((2-Fluorobenzyl)oxy)-3-methoxybenzaldehyde (2a)
2.3.2. 4-((2-Chlorobenzyl)oxy)-3-methoxybenzaldehyde (2b)
2.3.3. 4-((4-Chlorobenzyl)oxy)-3-methoxybenzaldehyde (2c)
2.4. General Synthesis of 4-Arylmethylen-2-pyrrolin-5-ones 3
2.4.1. Methyl (Z)-1-Benzyl-4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (3a)
2.4.2. Methyl (Z)-1-Benzyl-4-(4-methoxybenzylidene)-2-methyl-5-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (3b)
2.4.3. Methyl (Z)-1-Benzyl-4-(4-chlorobenzylidene)-2-methyl-5-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (3c)
2.4.4. Methyl (Z)-4-(Benzo[d](1,3)dioxol-5-ylmethylene)-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3d)
2.4.5. Methyl (Z)-4-(2,4-Dimethoxybenzylidene)-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3e)
2.4.6. Methyl (Z)-2-Methyl-5-oxo-1-phenethyl-4-(2,4,5-trimethoxybenzylidene)-4,5-dihydro-1H-pyrrole-3-carboxylate (3f)
2.4.7. Methyl (Z)-2-Methyl-5-oxo-1-phenethyl-4-(3,4,5-trimethoxybenzylidene)-4,5-dihydro-1H-pyrrole-3-carboxylate (3g)
2.4.8. Methyl (Z)-4-(3-Hydroxy-4-methoxybenzylidene)-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3- carboxylate (3h)
2.4.9. Methyl (Z)-4-(3,4-Dihydroxybenzylidene)-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3i)
2.4.10. Methyl (Z)-4-[4-((2-Fluorobenzyl)oxy)-3-methoxybenzylidene]-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3j)
2.4.11. Methyl (Z)-4-[4-((2-Chlorobenzyl)oxy)-3-methoxybenzylidene]-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3k)
2.4.12. Methyl (Z)-4-[4-((4-Chlorobenzyl)oxy)-3-methoxybenzylidene]-2-methyl-5-oxo-1-phenethyl-4,5-dihydro-1H-pyrrole-3-carboxylate (3l)
2.4.13. Methyl (Z)-4-[4-((4-Chlorobenzyl)oxy)-3-methoxybenzylidene]-1-(3,4-dimethoxyphenethyl)-2-methyl-5-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (3m)
2.4.14. Methyl (Z)-4-[4-((2-Chlorobenzyl)oxy)-3-methoxybenzylidene]-1-(3,4-dimethoxyphenethyl)-2-methyl-5-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (3n)
2.4.15. Methyl (Z)-4-((1H-Indol-3-yl)methylene)-2-methyl-5-oxo-1-phenethyl-4,5- dihydro-1H-pyrrole-3-carboxylate (3o)
2.5. Antioxidant Assesment by the 1,1-Diphenyl-2-picryl-hydrazyl (DPPH) Method
2.6. Antioxidant Assesment by the Ferric Reducing Antioxidant Power (FRAP) Method
2.7. Acetylcholinesterase (AChE) Inhibition Assay
2.8. Determination of Nrf2 Transcription Factor Induction
2.9. Immunocytochemistry
2.10. Western Blot Analysis
2.11. Culture of SH-SY5Y Cells
2.12. SH-SY5Y In Vitro Neuroprotection Studies
2.13. Reactive Oxygen Species Production
2.14. Superoxide Production Measurement
2.15. Statistical Analysis
3. Results
3.1. Synthesis of Bisavenantramide Analogs 3
3.2. Computational Druggability Study of Compounds 3
3.3. Characterization of Bisavenantramide Analogs 3 as Antioxidants
3.4. Acetylcholinesterase (AChE) Inhibitory Activity
3.5. Biological Evaluation of Bisavenantramide Analogues
3.5.1. Cytotoxicity Evaluation in the SHSY5Y Cell Line
3.5.2. Nrf2 Induction
3.5.3. Compound 3g Induces Nrf2 Nuclear Translocation and Nrf2-ARE Dependant Protein Expression
3.5.4. Neuroprotection in a Rotenone/Oligomycin A Oxidative Stress Model
3.5.5. Neuroprotection against Tau Hyperphosphorylation Induced by Okadaic Acid
3.5.6. Compound 3i Affords Neuroprotection by Reducing Free Radical Production and Tau Hyperphosphorylation and also by Nrf2 Induction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Kim, G.A.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 34, 325–340. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Koppula, S.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Nuclear factor erythroid 2-related factor 2 signaling in Parkinson disease: A promising multi therapeutic target against oxidative stress, neuroinflammation and cell death. CNS Neurol. Disord. Drug Targets 2012, 11, 1015–1029. [Google Scholar] [CrossRef]
- Cores, A.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. Nrf2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the Nrf2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317, and references therein. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Kirberger, M.; Yang, Y.; Chen, N. Natural products for cancer prevention associated with Nrf2–ARE pathway. Food Sci. Hum. Wellness 2013, 2, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Michaluart, P.; Masferrer, J.L.; Carothers, A.M.; Subbaramaiah, K.; Zweifel, B.S.; Koboldt, C.; Mestre, J.R.; Grunberger, D.; Sacks, P.G.; Tanabe, T.; et al. Inhibitory effects of caffeic acid phenethyl ester on the activity and expression of cyclooxygenase-2 in human oral epithelial cells and in a rat model of inflammation. J. Cancer Res. 1999, 59, 2347–2352. [Google Scholar]
- Scapagnini, G.; Butterfield, D.A.; Colombrita, C.; Sultana, R.; Pascale, A.; Calabrese, V. Ethyl ferulate, a lipophilic polyphenol, induces HO-1 and protects rat neurons against oxidative stress. Antioxid. Redox. Signal 2004, 6, 811–818. [Google Scholar] [PubMed]
- Yang, J.; Ou, B.X.; Wise, M.L.; Chu, Y.F. In vitro total antioxidant capacity and anti-inflammatory activity of three common oat-derived avenanthramides. Food Chem. 2014, 160, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhu, Y.; Yerke, A.; Wise, M.L.; Johnson, J.; Chu, Y.; Sang, S. Oat avenanthramides induce heme oxygenase-1 expression via Nrf2-mediated signaling in HK-2 cells. Mol. Nutr. Food Res. 2015, 59, 2471–2479. [Google Scholar] [CrossRef]
- Di Maso, M.J.; Nepomuceno, G.M.; St. Peter, M.A.; Gitre, H.H.; Martin, K.S.; Shaw, J.T. Synthesis of (±)-bisavenanthramide B-6 by an anionic anhydride Mannich reaction. Org. Lett. 2016, 18, 1740–1743. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Ishizuka, A.; Ishihara, A.; Nishioka, T.; Iwamura, H. New dimeric compounds of avenanthramide phytoalexin in oats. J. Org. Chem. 2007, 72, 3830–3839. [Google Scholar] [CrossRef] [PubMed]
- Cores, A. Natural Product-Related Multitarget-Directed Ligands for the Potential Treatment of Neurodegenerative Diseases. Ph.D. Thesis, Universidad Complutense, Madrid, Spain, 2019. [Google Scholar]
- Cores, A.; Estévez, V.; Villacampa, M.; Menéndez, J.C. Three-component access to 2-pyrrolin-5-ones and their use in target-oriented and diversity-oriented synthesis. RSC Adv. 2016, 6, 39433–39443. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Tetz, L.M.; Kamau, P.W.; Cheng, A.A.; Meeker, J.D.; Loch-Caruso, R. Troubleshooting the dichlorofluorescein assay to avoid artifacts in measurement of toxicant-stimulated cellular production of reactive oxidant species. J. Pharmacol. Toxicol. Methods 2013, 67, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martínez-González, L.; Roca, C.; Campillo, N.E.; Zaldívar-Díez, J.; Pérez, C.; Zuccheri, G.; Miti, A.; et al. Tau-centric multitarget approach for Alzheimer’s disease: Development of first-in-class dual glycogen synthase kinase 3β and tau-aggregation inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ácsová, A.; Martiniaková, S.; Hojerová, J. Selected in vitro methods to determine antioxidant activity of hydrophilic/lipophilic substances. Acta Chim. Slovaca 2019, 12, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, F.; Konstantinidou, M.; Dömling, M. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Bian, Y.; Jungho, J.J.; Cuyler, J.; Xi, X.-Q. Covalent allosteric modulation: An emerging strategy for GPCRs drug discovery. Eur. J. Med. Chem. 2020, 206, 1126902. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element–driven reporter gene cell line and its use to show redox-dependent activation of Nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; McDonald, P.; Liu, J.; Klaassen, C. Screening of natural compounds as activators of the keap1-nrf2 pathway. Planta Medica 2013, 80, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, I.; Navarro, E.; Michalska, P.; Gameiro, I.; Egea, J.; Abril, S.; López, A.; González-Lafuente, L.; López, M.G.; León, R. New melatonin–cinnamate hybrids as multi-target drugs for neurodegenerative diseases: Nrf2-induction, antioxidant effect and neuroprotection. Future Med. Chem. 2015, 7, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Tenti, G.; Parada, E.; León, R.; Egea, J.; Martínez-Revelles, S.; Briones, A.M.; Martínez-Revelles, S.; Briones, A.M.; Sridharan, V.; López, M.G.; et al. New 5-unsubstituted dihydropyridines with improved CaV1. 3 selectivity as potential neuroprotective agents against ischemic injury. J. Med. Chem. 2014, 57, 4313–4323. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, T.J. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Xiong, N.; Xiong, J.; Jia, M.; Liu, L.; Zhang, X.; Chen, Z.; Huang, J.; Zhang, Z.; Hou, L.; Luo, Z.; et al. The role of autophagy in Parkinson’s disease: Rotenone-based modeling. Behav. Brain Funct. 2013, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Almghandi, B.S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau pathology in Parkinson’s disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloret, A.; Badia, M.-C.; Giraldo, E.; Ermak, G.; Alonso, M.-D.; Pallardó, F.V.; Davies, K.E.; Viña, J. Amyloid-toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Comp. | R1 | R2 | Yield, % |
|---|---|---|---|---|
| 1 | 3a | C6H5CH2 | H | 50 |
| 2 | 3b | C6H5CH2 | 4-OMe | 60 |
| 3 | 3c | C6H5CH2 | 4-Cl | 45 |
| 4 | 3d | C6H5CH2CH2 | 3,4-OCH2O | 60 |
| 5 | 3e | C6H5CH2CH2 | 2,4-(MeO)2 | 41 |
| 6 | 3f | C6H5CH2CH2 | 2,4,5-(MeO)3 | 47 |
| 7 | 3g | C6H5CH2CH2 | 3,4,5-(MeO)3 | 53 |
| 8 | 3h | C6H5CH2CH2 | 3-OH, 4-OMe | 36 |
| 9 | 3i | C6H5CH2CH2 | 3,4-(OH)2 | 24 |
| 10 | 3j | C6H5CH2CH2 | 3-OMe, 4-(2-FC6H4CH2O) | 28 |
| 11 | 3k | C6H5CH2CH2 | 3-OMe, 4-(2-ClC6H4CH2O) | 37 |
| 12 | 3l | C6H5CH2CH2 | 3-OMe, 4-(4-ClC6H4CH2O) | 38 |
| 13 | 3m | 3,4(MeO)2C6H3CH2CH2 | 3-OMe, 4-(4-ClC6H4CH2O) | 43 |
| 14 | 3n | 3,4(MeO)2C6H3CH2CH2 | 3-OMe, 4-(2-ClC6H4CH2O) | 46 |
| Entry | Compound | DPPH | FRAP | |||
|---|---|---|---|---|---|---|
| Scavenging at 0.1 mM, % | Scavenging at 1 mM, % | IC50, μM | nmol Fe2+ at 30 µM | TEAC | ||
| 1 | Trolox | 11.4 ± 1.0 (9) | 2.28 ± 0.15 (9) | 1.00 | ||
| 2 | Ascorbic acid | 16.2 ± 0.7 (9) | ||||
| 3 | Melatonin | 1988 ± 1397 (2) | ||||
| 4 | Ferulic acid | 1.98 ± 0.04 (3) | 0.87 | |||
| 5 | 3a | 9.7 ± 3.8 | 47.5 ± 4.9 | - | 1.41 ± 0. 03 (3) | 0.62 |
| 6 | 3b | 6.5 ± 4.5 | 27.2 ± 6.4 | - | 1.44 ± 0.02 (3) | 0.63 |
| 7 | 3c | 3.7 ± 3.8 | 36.6 ± 3.4 | - | 1.41 ± 0.03 (3) | 0.62 |
| 8 | 3d | 40.5 ± 5.0 | 41.6 ± 4.1 | - | 1.41 ± 0.04 (3) | 0.62 |
| 9 | 3e | 6.33 ± 4.2 | 27.0 ± 2.6 | - | 1.44 ± 0.01 (3) | 0.63 |
| 10 | 3f | 9.7 ± 0.6 | 9.7 ± 2.2 | - | 1.42 ± 0.04 (3) | 0.62 |
| 11 | 3g | 8.1 ± 4.7 | 46.5 ± 2.3 | - | 1.36 ± 0.10 (3) | 0.60 |
| 12 | 3h | 89.0 ± 0.4 | - | 26.3 ± 2.1 (3) | 1.63 ± 0.04 (3) | 0.71 |
| 13 | 3i | 90.9 ± 0.5 | - | 7.5 ± 0.4 (3) | 2.30 ± 0.08 (3) | 1.01 |
| 14 | 3j | 4.3 ± 2.1 | 31.3 ± 3.3 | - | 1.60 ± 0.04 (3) | 0.70 |
| 15 | 3k | 7.3 ±1.4 | 27.3 ± 1.0 | - | 1.63 ± 0.01 (3) | 0.71 |
| 16 | 3l | 11.3 ± 2.6 | 63.5 ± 2.2 | 712.2 ± 5.4 (3) | 1.62 ± 0.03 (3) | 0.71 |
| 17 | 3m | 22.8 ± 0.2 | 85.5 ± 2.7 | 334.3 ± 22.9 (3) | 1.59 ± 0.05 (3) | 0.70 |
| 18 | 3n | 12.7 ± 3.5 | 73.6 ± 0.7 | 552.1 ± 17.4 (3) | 1.59 ± 0.02 (3) | 0.69 |
| 19 | 3o | 12.7 ± 1.9 | 49.9 ± 3.4 | - | 1.80 ± 0.07 (3) | 0.79 |
| Entry | Compound | % Inhibition of EeAChE at 10 μM | IC50 (μM) |
|---|---|---|---|
| 1 | 3a | 27.36 | |
| 2 | 3b | 36.71 | 29.7 ± 3.0 (3) |
| 3 | 3c | 33.47 | |
| 4 | 3d | 25.21 | |
| 5 | 3e | 24.32 | |
| 6 | 3f | 29.29 | |
| 7 | 3g | 11.62 | |
| 8 | 3h | 24.25 | |
| 9 | 3i | 27.21 | |
| 10 | 3j | 15.96 | |
| 11 | 3k | 40.09 | 22.4 ± 3.2 (3) |
| 12 | 3l | 26.20 | |
| 13 | 3m | 31.77 | |
| 14 | 3n | 17.59 | |
| 15 | 3o | 39.84 | 12.5 ± 1.4 (3) |
| Entry | Compound | LD50 (μM) |
|---|---|---|
| 1 | 3a | 63.7 ± 12.8 |
| 2 | 3b | >100 |
| 3 | 3c | 75.4 ± 1 5.6 |
| 4 | 3d | >100 |
| 5 | 3e | >100 |
| 6 | 3f | >100 |
| 7 | 3g | >100 |
| 8 | 3h | >100 |
| 9 | 3i | 92.8 ± 4.6 |
| 10 | 3j | >100 |
| 11 | 3k | >100 |
| 12 | 3l | >100 |
| 13 | 3m | >100 |
| 14 | 3n | >100 |
| 15 | 3o | >100 |
| Entry | Compound | CD (μM) |
|---|---|---|
| 1 | Caffeic acid | 6.5 to 11.75 |
| 2 | 3a | 30.22 ± 1.89 |
| 3 | 3b | 17.96 ± 1.84 |
| 4 | 3c | 15.00 ± 0.80 |
| 5 | 3d | 17.86 ± 8.58 |
| 6 | 3e | 10.38 ± 0.80 |
| 7 | 3f | 31.06 ± 6.21 |
| 8 | 3g | 3.18 ± 1.16 |
| 9 | 3h | 36.07 ± 418 |
| 10 | 3i | 7.51 ± 3.45 |
| 11 | 3j | 11.91 ± 2.83 |
| 12 | 3k | 7.58 ± 4.35 |
| 13 | 3l | 13.97 ± 4.45 |
| 14 | 3m | 21.41 ± 5.72 |
| 15 | 3n | 14.26 ± 3.89 |
| 16 | 3o | 35.98 ± 10.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cores, Á.; Abril, S.; Michalska, P.; Duarte, P.; Olives, A.I.; Martín, M.A.; Villacampa, M.; León, R.; Menéndez, J.C. Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants 2021, 10, 941. https://doi.org/10.3390/antiox10060941
Cores Á, Abril S, Michalska P, Duarte P, Olives AI, Martín MA, Villacampa M, León R, Menéndez JC. Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants. 2021; 10(6):941. https://doi.org/10.3390/antiox10060941
Chicago/Turabian StyleCores, Ángel, Sheila Abril, Patrycja Michalska, Pablo Duarte, Ana I. Olives, M. Antonia Martín, Mercedes Villacampa, Rafael León, and J. Carlos Menéndez. 2021. "Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation" Antioxidants 10, no. 6: 941. https://doi.org/10.3390/antiox10060941
APA StyleCores, Á., Abril, S., Michalska, P., Duarte, P., Olives, A. I., Martín, M. A., Villacampa, M., León, R., & Menéndez, J. C. (2021). Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants, 10(6), 941. https://doi.org/10.3390/antiox10060941