
Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
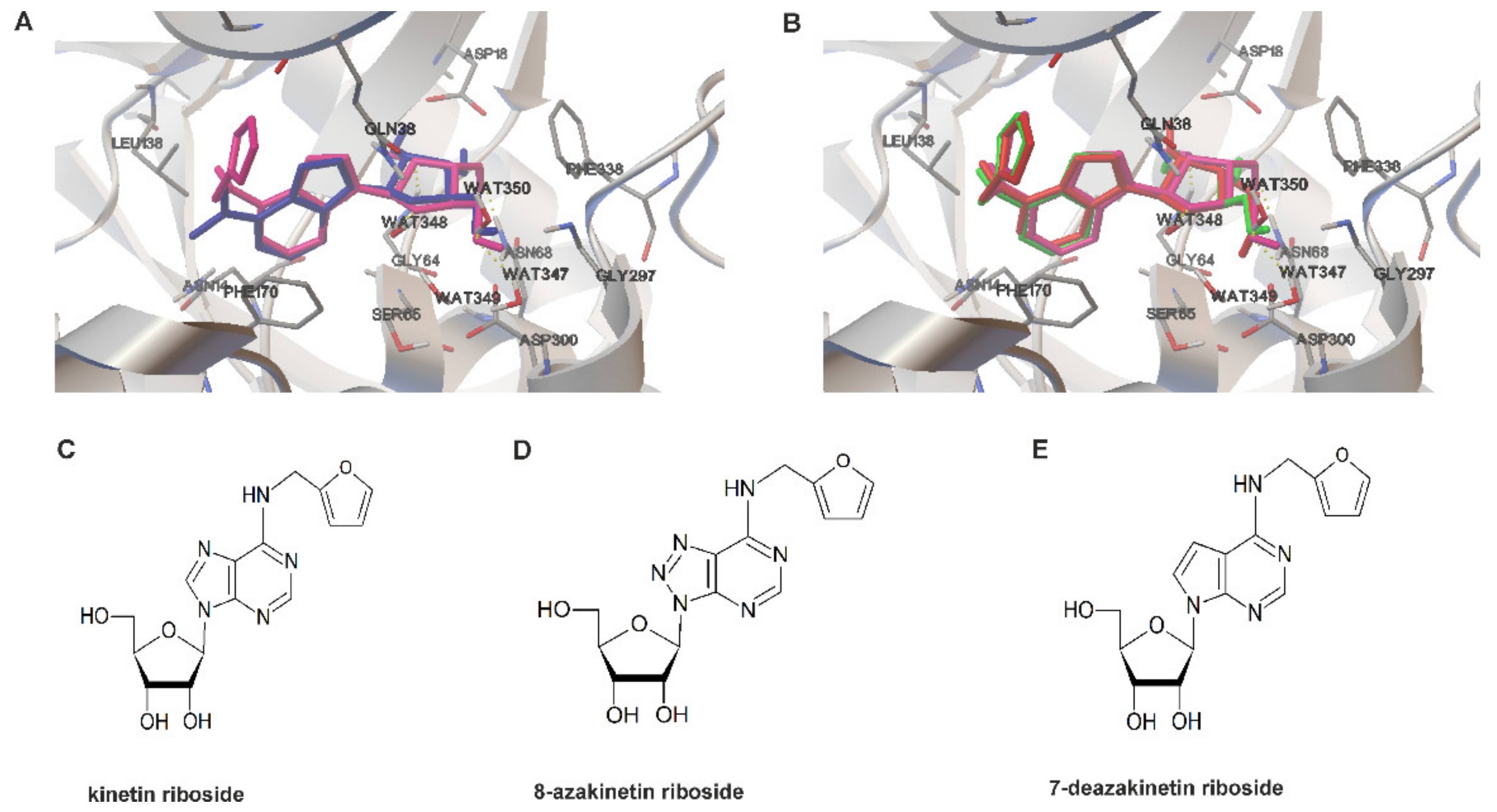
2.2. Homology Modeling of Semi-Open Human Adenosine Kinase Conformation
2.3. Ligand Preparation and Molecular Docking
2.4. Synthesis of 8-azaKR and 7-deazaKR
2.5. Cell Culture
2.6. Oxygen Consumption Analysis
2.7. Comparative Mitochondrial ROS Analysis by Flow Cytometry
2.8. Spheroid Formation, Treatment, and Labeling Preparation for Screening of KR Derivatives
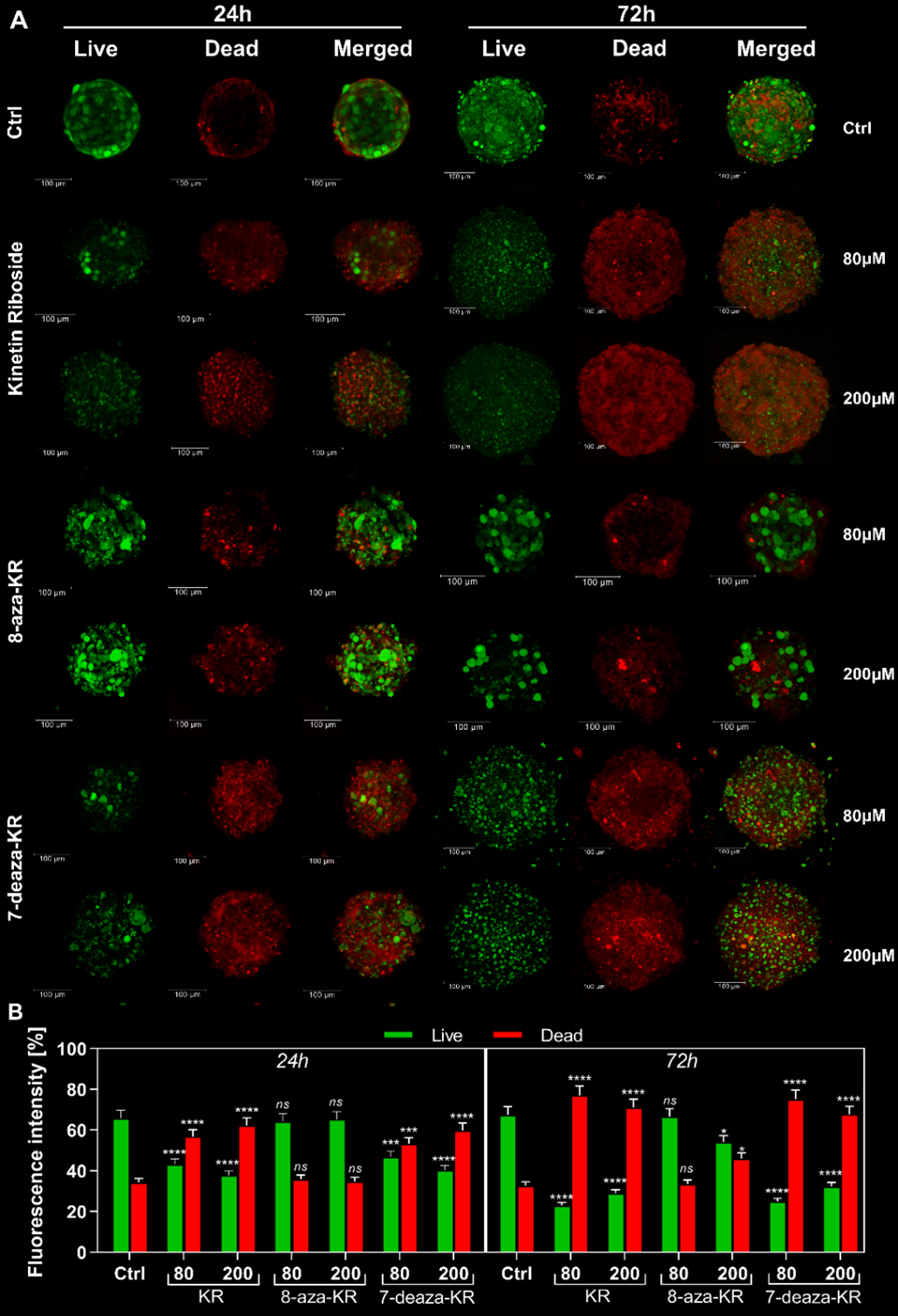
2.8.1. LIVE/DEAD Analysis of T98G Spheroids
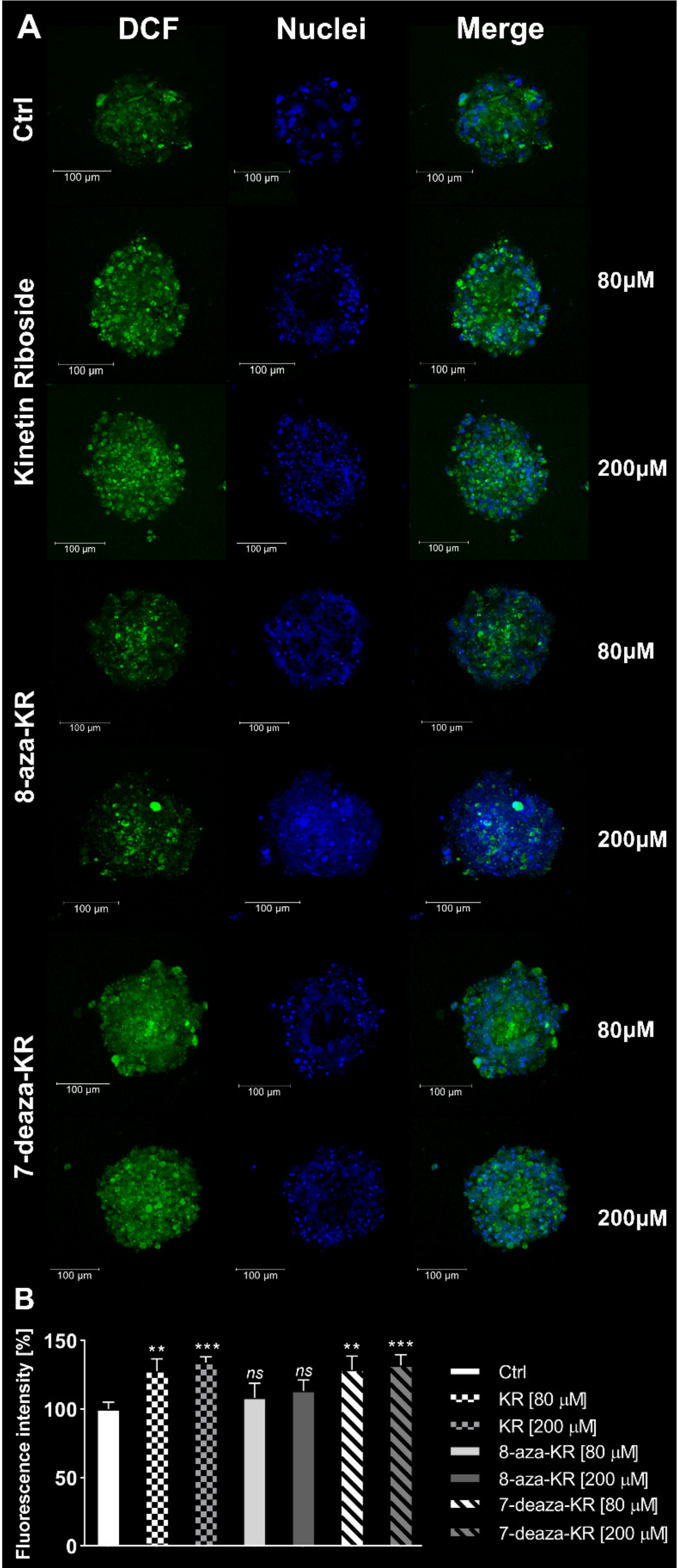
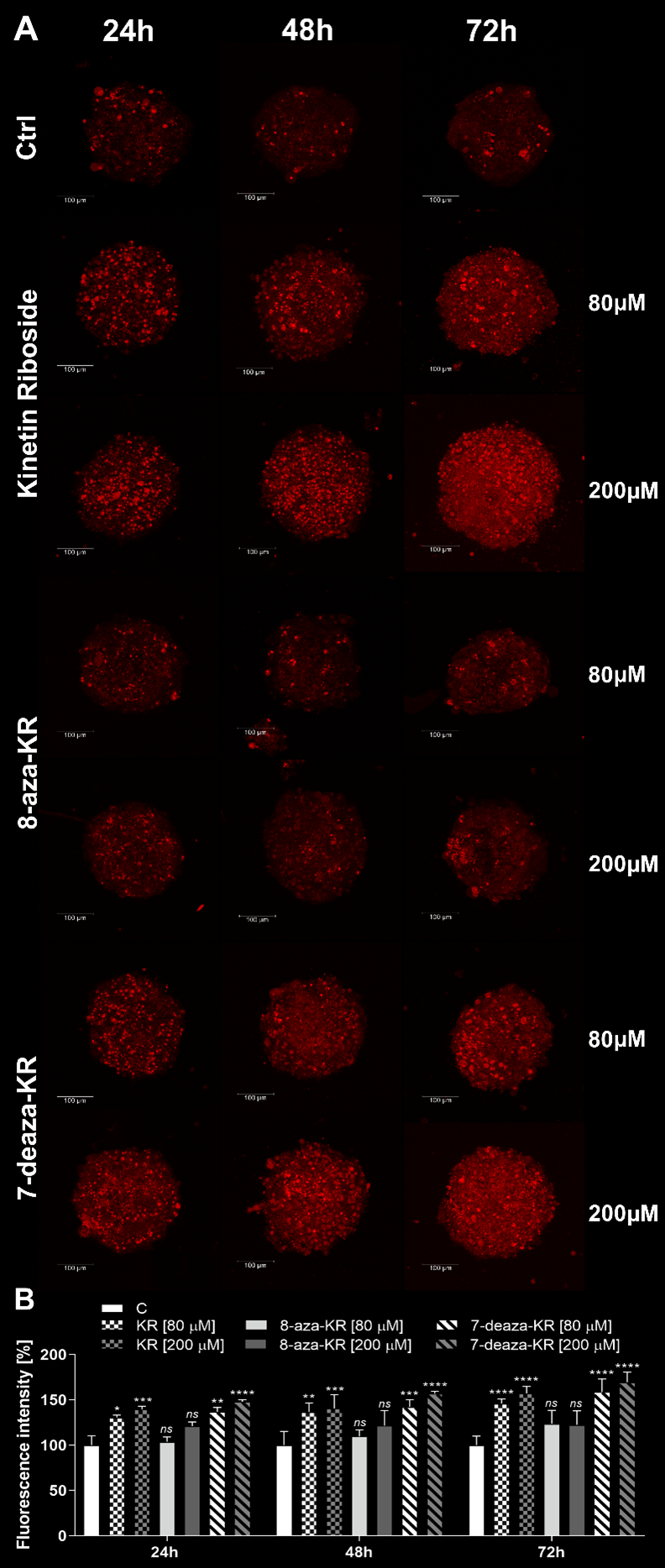
2.8.2. Intracellular and Mitochondrial Oxidative Stress Measurements in T98G Spheroids
2.9. Comparative Analysis of the Anticancer and Pro-Oxidative Properties of KR and 7-deazaKR by Using Two-Dimensional Culture of T98G Cells
2.9.1. In Vitro Phosphorylation
2.9.2. Bioluminescent Measurement of Cellular ATP Content
2.9.3. Real-Time Analysis of Cell Proliferation Using the xCELLigence System
2.9.4. Apoptosis/Necrosis Assay by Flow Cytometry
2.9.5. Metabolic OS (Intracellular and Mitochondrial) Measurement by Flow Cytometry
2.9.6. Lipid Peroxidation Measurements by Flow Cytometry
2.9.7. Glutathione Level Measurement by Flow Cytometry
2.9.8. Analysis of 8-oxo-dG in T98G Cells by HPLC-UV-ED
2.9.9. Total RNA Isolation
2.9.10. cDNA Synthesis and Real-Time
2.10. Statistical Analysis
3. Results
3.1. Small Compounds as a Part of Oxidative Therapy in GBM Cells: Metabolic Profiles and Comparative Analysis of Mitochondrial ROS in T98G vs. HepG2 Cells
3.2. Determination of KR Analogues Showing Similar Affinity for ADK
3.3. Impact of KR, 8-azaKR, and 7-deazaKR Treatment on the Viability of T98G Spheroids
3.4. Induction of the Intracellular and Mitochondrial OS in T98G Spheroids after Treatment with KR, 8-azaKR, and 7-deazaKR
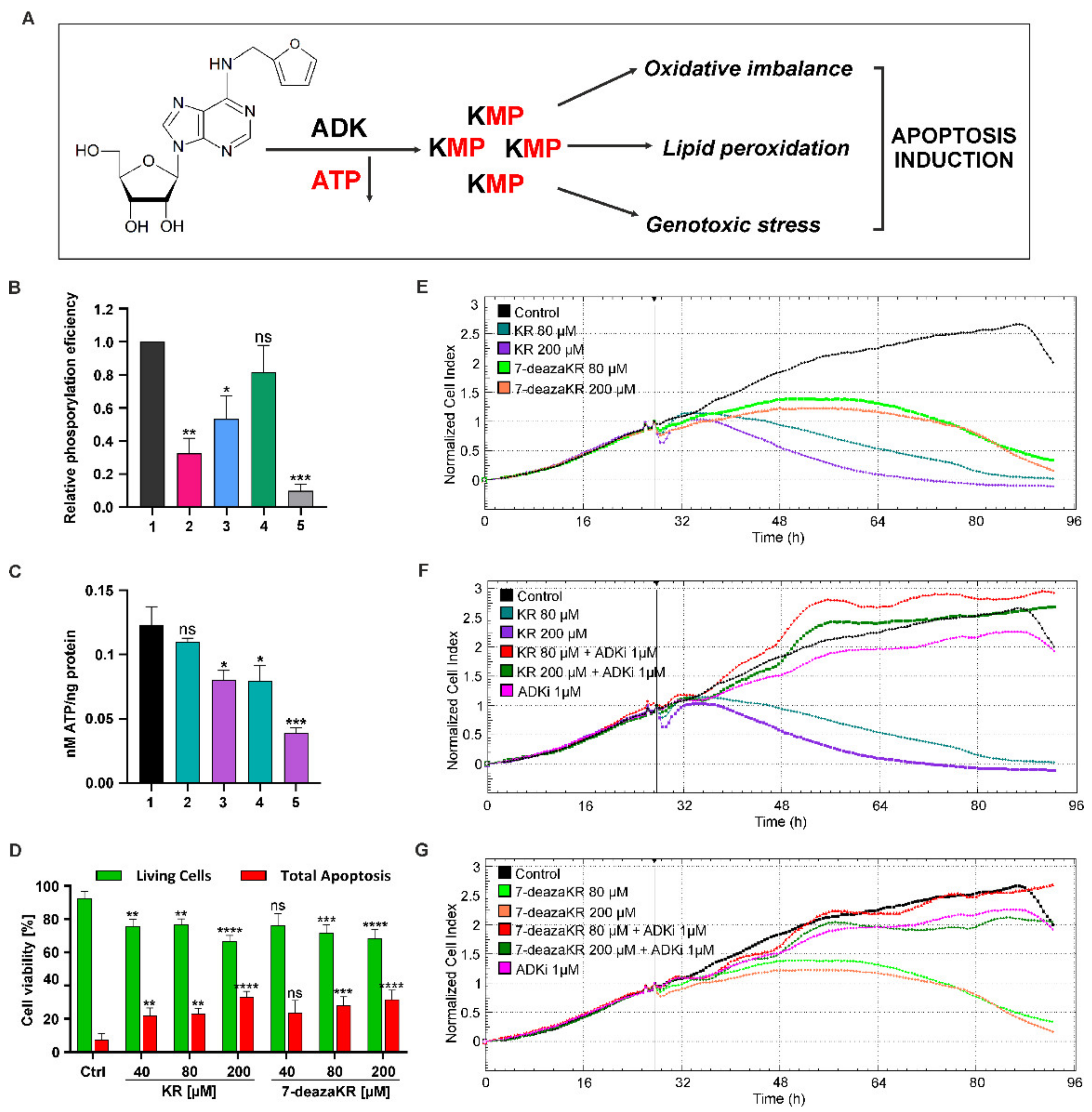
3.5. ADK Is Required for Complete Activity and Toxicity of the Adenosine Derivative through the Salvage Pathway. Treatment with KR and 7-deazaKR Induces Rapid Depletion of Cellular ATP Levels, Leading to T98G Cell Death
3.6. Disruption of the Oxidative Parameters in T98G Cells after KR and 7-deazaKR Treatment: The Effect of the Compounds on the Activation of Metabolic OS and Antioxidant Defense Mechanism
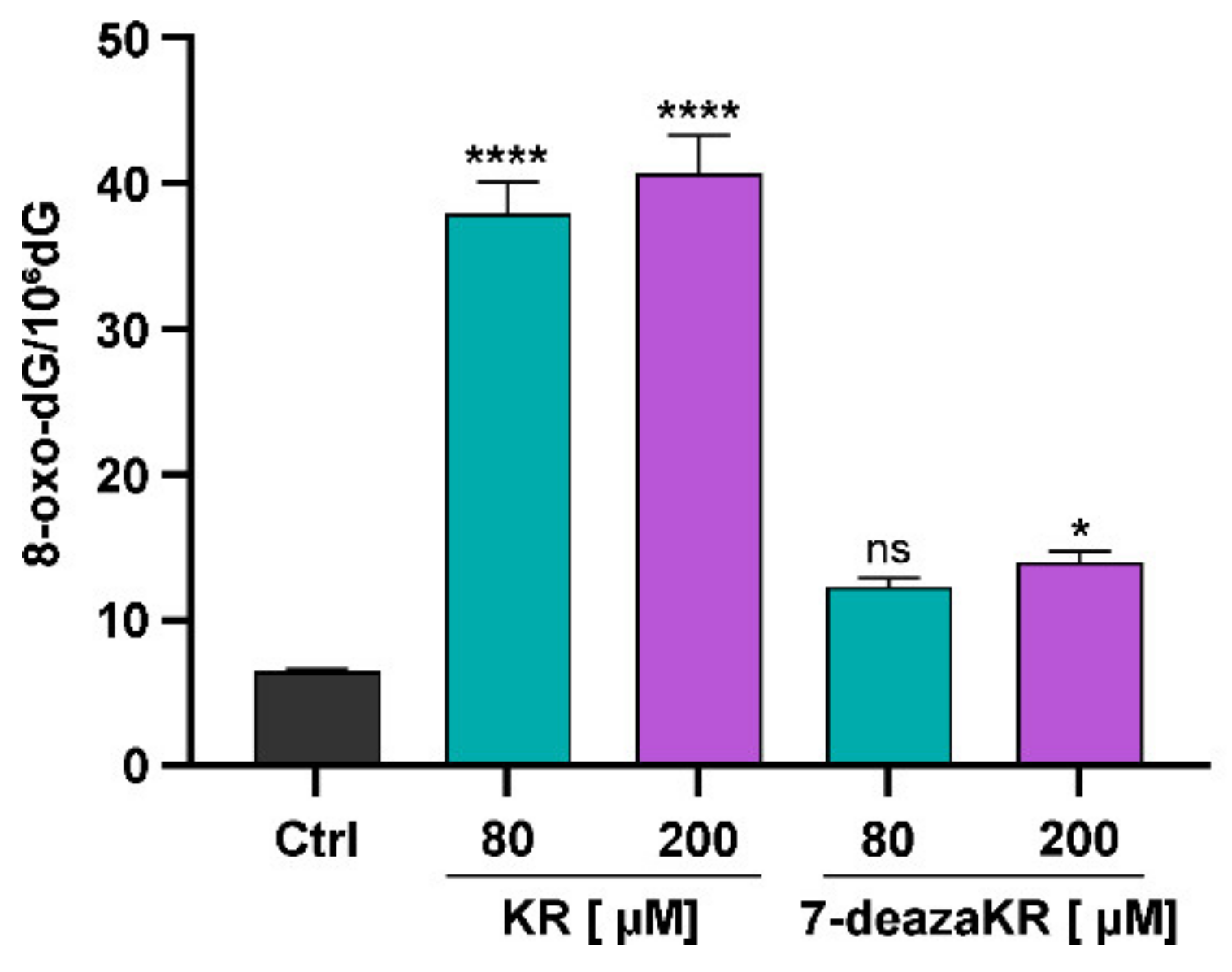
3.7. Effect of KR and 7-deazaKR on DNA Oxidation
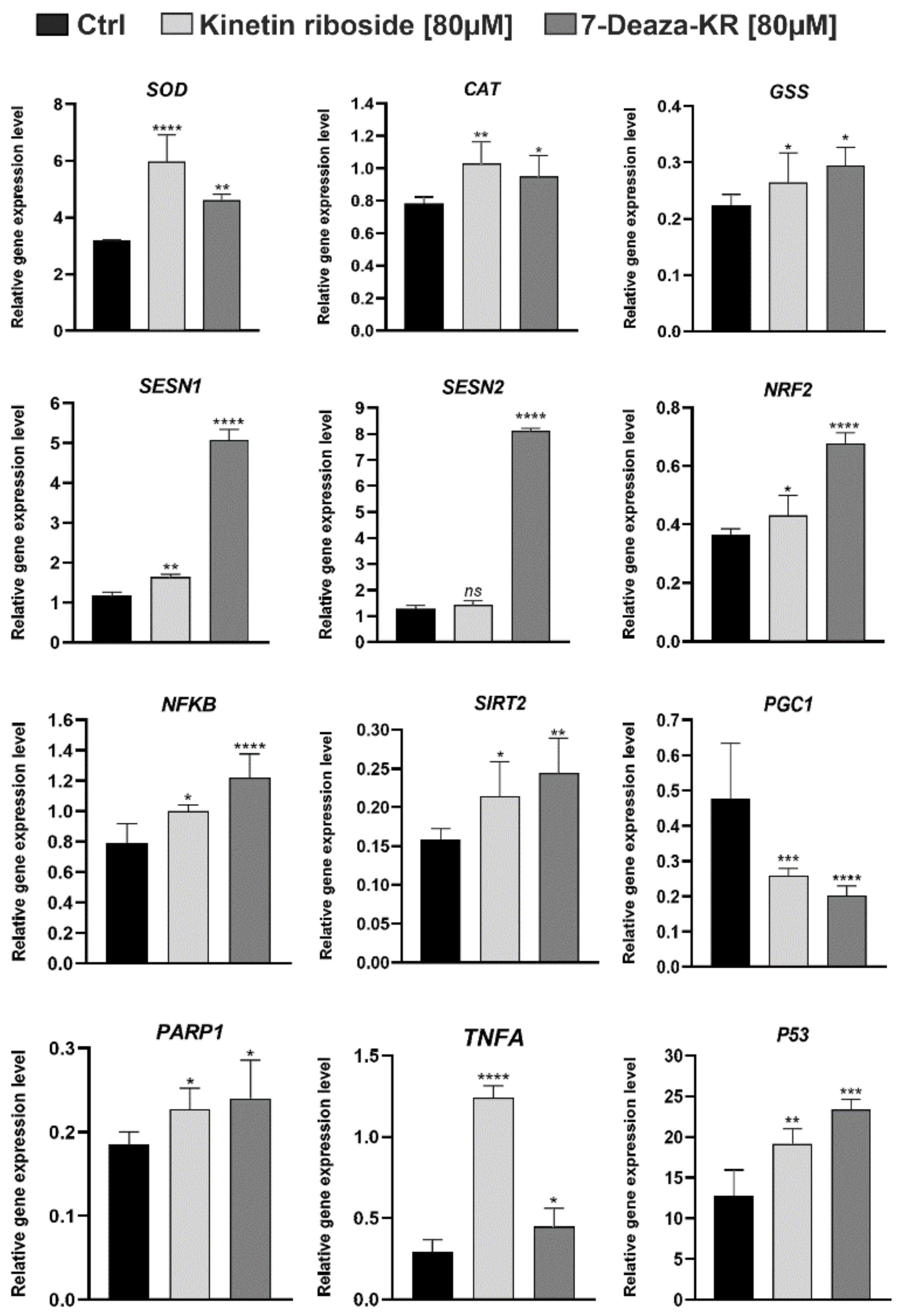
3.8. Effect of KR and 7-deazaKR on the Expression Level of Genes Involved in Oxidative Stress Response in T98G Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Taylor, O.G.; Brzozowski, J.; Skelding, K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [Green Version]
- Baillie, N.; Carr, A.C.; Peng, S. The use of intravenous vitamin C as a supportive therapy for a patient with glioblastoma multiforme. Antioxidants 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, B.; Zheng, X.; Wu, M.-L.; Tian, X.-T.; Song, X.; Liu, Y.-N.; Li, P.-N.; Liu, J. Increased reactive oxygen species and distinct oxidative damage in resveratrol-suppressed glioblastoma cells. J. Cancer 2021, 12, 141–149. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, Z.; Ding, Y.; Xue, K.; Wang, X.; Yang, R.; An, Y.; Liu, D.; Hu, C.; Tang, Q. LRP1-mediated pH-sensitive polymersomes facilitate combination therapy of glioblastoma in vitro and in vivo. J. Nanobiotechnol. 2021, 19, 29. [Google Scholar] [CrossRef]
- Bahadur, S.; Sahu, A.K.; Baghel, P.; Saha, S. Current promising treatment strategy for glioblastoma multiform: A review. Oncol. Rev. 2019, 13, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA Neurol. 2014, 71, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.K.; Sulman, E.P.; Wen, P.Y.; Kurz, S.C. Novel therapies for glioblastoma. Curr. Neurol. Neurosci. Rep. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.; Grześkowiak, B.F.; Szutkowski, K.; Wawrzyniak, D.; Głodowicz, P.; Barciszewski, J.; Jurga, S.; Rolle, K.; Mrówczyński, R. Nano-mediated delivery of double-stranded RNA for gene therapy of glioblastoma multiforme. PLoS ONE 2019, 14, e0213852. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B. Temozolomide in glioblastoma therapy: Role of apoptosis, senescence and autophagy. Comment on Strobel et al., Temozolomide and other alkylating agents in glioblastoma therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; De La Cruz, V.P.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of redox status in development of glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Krawczynski, K.; Godlewski, J.; Bronisz, A. Oxidative stress—Part of the solution or part of the problem in the hypoxic environment of a brain tumor. Antioxidants 2020, 9, 747. [Google Scholar] [CrossRef] [PubMed]
- Orlicka-Plocka, M.; Gurda-Wozna, D.; Fedoruk-Wyszomirska, A.; Wyszko, E. Circumventing the Crabtree effect: Forcing oxidative phosphorylation (OXPHOS) via galactose medium increases sensitivity of HepG2 cells to the purine derivative kinetin riboside. Apoptosis 2020, 25, 835–852. [Google Scholar] [CrossRef]
- Voller, J.; Zatloukal, M.; Lenobel, R.; Dolezal, K.; Béreš, T.; Krystof, V.; Spíchal, L.; Niemann, P.; Džubák, P.; Hajduch, M.; et al. Anticancer activity of natural cytokinins: A structure–activity relationship study. Phytochemistry 2010, 71, 1350–1359. [Google Scholar] [CrossRef]
- Zhang, Y.; El Kouni, M.H.; Ealick, S.E. Substrate analogs induce an intermediate conformational change in Toxoplasma gondii adenosine kinase. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Bhutoria, S.; Ghoshal, N. Deciphering ligand dependent degree of binding site closure and its implication in inhibitor design: A modeling study on human adenosine kinase. J. Mol. Graph. Model. 2010, 28, 577–591. [Google Scholar] [CrossRef]
- Mathews, I.I.; Erion, M.D.; Ealick, S.E. Structure of human adenosine kinase at 1.5 A resolution. Biochemistry 1998, 37, 15607–15620. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Lemmon, G.; Meiler, J. Towards ligand docking including explicit interface water molecules. PLoS ONE 2013, 8, e67536. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2010, 7, 525–537. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. Amber 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Baranowski, D.; Framski, G.; Wyszko, E.; Ostrowski, T. Studies on structure of kinetin riboside and its analogues by variable-temperature NMR. J. Mol. Struct. 2019, 1195, 110–118. [Google Scholar] [CrossRef]
- Barciszewska, A.-M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef] [Green Version]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug resistance in glioblastoma: The two faces of oxidative stress. Front. Mol. Biosci. 2021, 7. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, J.A.; Shortnacy, A.T.; Secrist, J.A. Synthesis and biological evaluation of 2-fluoro-8-azaadenosine and related-compounds. J. Med. Chem. 1983, 26, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Hardesty, C.T.; Chaney, N.A.; Waravdekar, V.S.; Mead, J.A. The disposition of the antitumor agent, sangivamycin, in mice. Cancer Res. 1974, 34, 1005–1009. [Google Scholar] [PubMed]
- Salem, M.A.I.; Marzouk, M.I.; El-Kazak, A.M. Synthesis and characterization of some new coumarins with in vitro antitumor and antioxidant activity and high protective effects against DNA damage. Molecules 2016, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z. Increased oxidative stress as a selective anticancer therapy. Oxidative Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.; Altavilla, D.; et al. ROS and brain gliomas: An overview of potential and innovative therapeutic strategies. Int. J. Mol. Sci. 2016, 17, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, P.R.D.V.; Pour Khavari, A.; Rizzo, M.; Sakamoto-Hojo, E.T.; Haghdoost, S. Targeting NRF2, regulator of antioxidant system, to sensitize glioblastoma neurosphere cells to radiation-induced oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 1–17. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [Green Version]
- Wyszko, E.; Rolle, K.; Nowak, S.; Zukiel, R.; Nowak, M.; Piestrzeniewicz, R.; Gawrońska, I.; Barciszewska, M.Z.; Barciszewski, J. A multivariate analysis of patients with brain tumors treated with ATN-RNA. Acta Pol. Pharm. Drug Res. 2009, 65, 677–684. [Google Scholar]
- Zukiel, R.; Nowak, S.; Wyszko, E.; Rolle, K.; Gawronska, I.; Barciszewska, M.Z.; Barciszewski, J. Suppression of human brain tumor with interference RNA specific for tenascin-C. Cancer Biol. Ther. 2006, 5, 1002–1007. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Braganhol, E.; Klafke, K.; Figueiró, F.; Terra, S.R.; Paludo, F.J.; Morrone, M.; Bristot, I.J.; Battastini, A.M.; Forcelini, C.M.; et al. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett. 2015, 358, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by which low glucose enhances the cyto-toxicity of metformin to cancer cells both in vitro and in vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiseleva, L.; Kartashev, A.V.; Vartanyan, N.L.; Pinevich, A.; Samoilovich, M.P. Characteristics of A172 and T98G cell lines. Tsitologiya 2016, 58, 349–355. [Google Scholar] [CrossRef]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007, 97, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Jelluma, N.; Yang, X.; Stokoe, D.; Evan, G.I.; Dansen, T.B.; Haas-Kogan, D.A. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006, 4, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Guntuku, L.; Naidu, V.; Yerra, V.G. Mitochondrial dysfunction in gliomas: Pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Song, Y.; Ren, Z.; Cheng, X.; Li, P.; Song, H.; Jia, L. Glioma cells are resistant to inflammation-induced alterations of mitochondrial dynamics. Int. J. Oncol. 2020, 57, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toler, S.M.; Noe, D.; Sharma, A. Selective enhancement of cellular oxidative stress by chloroquine: Implications for the treatment of glioblastoma multiforme. Neurosurg. Focus 2006, 21, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Barciszewska, A.-M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A new epigenetic mechanism of temozolomide action in glioma cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, T.V.; Lenz, L.S.; Bottari, N.B.; Pereira, A.; Schetinger, M.R.C.; Morsch, V.M.; Ulrich, H.; Pillat, M.M.; De Andrade, C.M. Berberine induces apoptosis in glioblastoma multiforme U87MG cells via oxidative stress and independent of AMPK activity. Mol. Biol. Rep. 2020, 47, 4393–4400. [Google Scholar] [CrossRef]
- Chiasserini, D.; Davidescu, M.; Orvietani, P.L.; Susta, F.; Macchioni, L.; Petricciuolo, M.; Castigli, E.; Roberti, R.; Binaglia, L.; Corazzi, L. 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteom. 2017, 152, 329–338. [Google Scholar] [CrossRef]
- Orlicka-Płocka, M.; Gurda, D.; Fedoruk-Wyszomirska, A.; Barciszewski, J.; Wyszko, E. Effect of kinetin riboside on proapoptotic activities in human cancer and normal cell lines. New Biotechnol. 2016, 33, S168. [Google Scholar] [CrossRef]
- Choi, B.H.; Kim, W.; Wang, Q.C.; Kim, D.C.; Tan, S.N.; Yong, J.W.; Kim, K.T.; Yoon, H.S. Kinetin riboside preferentially induces apoptosis by modulating Bcl-2 family proteins and caspase-3 in cancer cells. Cancer Lett. 2008, 261, 37–45. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., 3rd; Ley, S.; Lamore, S.D.; Azimian, S.; Wondrak, G.T. The experimental chemotherapeutic N6-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A(p21) upregulation in human cancer cell lines. Biochem. Pharmacol. 2009, 77, 1125–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudzik, P.; Dulińska-Litewka, J.; Wyszko, E.; Jędrychowska, P.; Opałka, M.; Barciszewski, J.; Laidler, P. Effects of kinetin riboside on proliferation and proapoptotic activities in human normal and cancer cell lines. J. Cell. Biochem. 2011, 112, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Ayres, M.L.; Kaluarachchi, K.; Plunkett, W.; Gandhi, V. Intracellular succinylation of 8-chloroadenosine and its effect on fumarate levels. J. Biol. Chem. 2010, 285, 8022–8030. [Google Scholar] [CrossRef] [Green Version]
- Santana, V.M.; Mirro, J.; Harwood, F.C.; Cherrie, J.; Schell, M.; Kalwinsky, D.; Blakley, R.L. A phase I clinical trial of 2-chlorodeoxyadenosine in pediatric patients with acute leukemia. J. Clin. Oncol. 1991, 9, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Stellrecht, C.M.; Vangapandu, H.V.; Le, X.-F.; Mao, W.; Shentu, S. ATP directed agent, 8-chloro-adenosine, induces AMP activated protein kinase activity, leading to autophagic cell death in breast cancer cells. J. Hematol. Oncol. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Rogalińska, M.; Góralski, P.; Błoński, J.Z.; Robak, P.; Barciszewski, J.; Koceva-Chyła, A.; Piekarski, H.; Robak, T.; Kilianska, Z.M. Personalized therapy tests for the monitoring of chronic lymphocytic leukemia development. Oncol. Lett. 2017, 13, 2079–2084. [Google Scholar] [CrossRef] [PubMed]
- Dulińska-Litewka, J.; Gąsiorkiewicz, B.; Litewka, A.; Gil, D.; Gołąbek, T.; Okoń, K. Could the kinetin riboside be used to inhibit human prostate cell epithelial–mesenchymal transition? Med. Oncol. 2020, 37, 17. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, J.; Shi, D.; Chen, W.; Li, J.; Yan, R.; Bi, Y.; Hu, W.; Zhu, Z.; Yu, Y.; et al. Glucocorticoids improve severe or critical COVID-19 by activating ACE2 and reducing IL-6 levels. Int. J. Biol. Sci. 2020, 16, 2382–2391. [Google Scholar] [CrossRef]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a type of three-dimensional cell cultures—Examples of methods of preparation and the most important application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dwarakanath, S.; Khaitan, D.; Chandna, S. Short-term exposure of multicellular tumor spheroids of a human glioma cell line to the glycolytic inhibitor 2-deoxy-D-glucose is more toxic than continuous exposure. J. Cancer Res. Ther. 2009, 5, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Łukasz; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Mao, X.; Shi, C.-X.; Zhu, Y.X.; Palmer, S.E.; Sebag, M.; Marler, R.; Chesi, M.; Fonseca, R.; Bergsagel, P.L.; et al. Identification of kinetin riboside as a repressor of CCND1 and CCND2 with preclinical antimyeloma activity. J. Clin. Investig. 2008, 118, 1750–1764. [Google Scholar] [CrossRef] [Green Version]
- Voller, J.; Béres, T.; Zatloukal, M.; Kaminski, P.A.; Niemann, P.; Dolezal, K.; Džubák, P.; Hajduch, M.; Strnad, M. The natural cytokinin 2OH3MeOBAR induces cell death by a mechanism that is different from that of the “classical” cytokinin ribosides. Phytochemistry 2017, 136, 156–164. [Google Scholar] [CrossRef]
- McDermott, S.P.; Eppert, K.; Notta, F.; Isaac, M.; Datti, A.; Al-Awar, R.; Wrana, J.; Minden, M.D.; Dick, J.E. A small molecule screening strategy with validation on human leukemia stem cells uncovers the therapeutic efficacy of kinetin riboside. Blood 2012, 119, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Delic, V.; Noble, K.; Zivkovic, S.; Phan, T.-A.; Reynes, C.; Zhang, Y.; Phillips, O.; Claybaker, C.; Ta, Y.; Dinh, V.B.; et al. The effects of AICAR and rapamycin on mitochondrial function in immortalized mitochondrial DNA mutator murine embryonic fibroblasts. Biol. Open 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Spitz, U.R.; Sim, J.E.; Ridnour, L.A.; Galoforo, S.S.; Lee, Y.J. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann. N. Y. Acad. Sci. 2006, 899, 349–362. [Google Scholar] [CrossRef]
- Qu, H.; Song, X.; Song, Z.; Jiang, X.; Gao, X.; Bai, L.; Wu, J.; Na, L.; Yao, Z. Berberine reduces temozolomide resistance by inducing autophagy via the ERK1/2 signaling pathway in glioblastoma. Cancer Cell Int. 2020, 20, 592. [Google Scholar] [CrossRef]
- Tong, L.; Xie, C.; Wei, Y.; Qu, Y.; Liang, H.; Zhang, Y.; Xu, T.; Qian, X.; Qiu, H.; Deng, H. Antitumor effects of berberine on gliomas via inactivation of caspase-1-mediated IL-1beta and IL-18 Release. Front. Oncol. 2019, 9, 364. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxidative Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2017, 144, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, H.; Park, S.; Yang, C.; Song, G.; Lim, W. Disruption of endoplasmic reticulum and ROS production in human ovarian cancer by campesterol. Antioxidants 2021, 10, 379. [Google Scholar] [CrossRef]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekvinda, J.; Różycka, D.; Rykowski, S.; Wyszko, E.; Fedoruk-Wyszomirska, A.; Gurda, D.; Orlicka-Płocka, M.; Giel-Pietraszuk, M.; Kiliszek, A.; Rypniewski, W.; et al. Synthesis of naphthalimide-carborane and metallacarborane conjugates: Anticancer activity, DNA binding ability. Bioorg. Chem. 2020, 94, 103432. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Mi, Y.; Qian, H.; Guo, N.; Yan, A.; Zhang, Y.; Gao, X. A potential mechanism of temozolomide resistance in glioma–ferroptosis. Front. Oncol. 2020, 10, 897. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, R.; Zhang, C. Erastin induces apoptotic and ferroptotic cell death by inducing ROS accumulation by causing mitochondrial dysfunction in gastric cancer cell HGC-27. Mol. Med. Rep. 2020, 22, 2826–2832. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.-L.; Wu, H.-T.; Chen, W.-J.; Xu, Y.; Ye, Q.-Q.; Shen, J.-X.; Liu, J. Involvement of glutathione peroxidases in the occurrence and development of breast cancers. J. Transl. Med. 2020, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting glutathione metabolism: Partner in crime in anticancer therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef] [Green Version]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Bak, A.; Kiliszek, A.; et al. Design, synthesis, and evaluation of novel 3-carboranyl-1,8-naphthalimide derivatives as potential anticancer agents. Int. J. Mol. Sci. 2021, 22, 2772. [Google Scholar] [CrossRef]
- Castro, M.L.; Carson, G.M.; McConnell, M.J.; Herst, P.M. High dose ascorbate causes both genotoxic and metabolic stress in glioma cells. Antioxidants 2017, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-Y.; Hsu, T.-I.; Hsu, C.-C.; Tsai, S.-Y.; Liu, J.-J.; Chou, S.-W.; Liu, M.-S.; Liou, J.-P.; Ko, C.-Y.; Chen, K.-Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O6-methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, S.-H.; Fay, J.; Cyuzuzo, C.I.; Jang, Y.; Lee, N.; Song, H.-S.; Harris, E.N. SESTRINs: Emerging dynamic stress-sensors in metabolic and environmental health. Front. Cell Dev. Biol. 2020, 8, 603421. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Narasimamurthy, R.; Xia, Y.; Myskiw, C.; Soda, Y.; Verma, I.M. Targeting NF-kappaB in glioblastoma: A therapeutic approach. Sci. Adv. 2016, 2, e1501292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawirska-Wojtasiak, R.; Fedoruk-Wyszomirska, A.; Piechowska, P.; Mildner-Szkudlarz, S.; Bajerska, J.; Wojtowicz, E.; Przygonski, K.; Gurda, D.; Kubicka, W.; Wyszko, E. Beta-carbolines in experiments on laboratory animals. Int. J. Mol. Sci. 2020, 21, 5245. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, S.-H.; Yi, M.-H.; Zhang, E.; Kim, E.; Park, J.; Jo, E.-K.; Lee, Y.H.; Park, M.S.; Kim, Y.; et al. Expression of PGC1α in glioblastoma multiforme patients. Oncol. Lett. 2017, 13, 4055–4076. [Google Scholar] [CrossRef] [Green Version]
- Griffaut, B.; Bos, R.; Maurizis, J.-C.; Madelmont, J.-C.; Ledoigt, G. Cytotoxic effects of kinetin riboside on mouse, human and plant tumour cells. Int. J. Biol. Macromol. 2004, 34, 271–275. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Full Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | UPL No. | |
|---|---|---|---|---|---|
| Control genes | ACTB | Actin Beta | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG | 64 |
| TBP | TATA-Box-Binding Protein | CGGCTGTTTAACTTCGCTTC | CACACGCCAAGAAACAGTGA | 3 | |
| PGK1 | Phosphoglycerate Kinase 1 | ACGCTACTGCATTCCTGCTT | ACTGTTTTGTGGGGTTTTTGTT | 13 | |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase 1 | TGACCTTGATTTATTTTGCATACC | CGAGCAAGACGTTCAGTCCT | 73 | |
| Target genes | CAT | Catalase | TCATCAGGGATCCCATATTGTT | CCTTCAGATGTGTCTGAGGATTT | 76 |
| GSS | Glutathione Synthetase | CCTGCTAGTGGATGCTGTCA | TCATCCTGTTTGATGGTGCT | 1 | |
| SOD | Superoxide Dismutase | TCCATGTTCATGAGTTTGGAGAT | TCTGGATAGAGGATTAAAGTGAGGA | 40 | |
| SESN1 | Sestrin 1 | GGGCCGTTACCCCTACATTA | TTCACTAAGTAGGAGCACTGATGTC | 46 | |
| SESN2 | Sestrin 2 | TCCGCCACTCAGAGAAGG | GGAGGGCGTACAGCAGAG | 68 | |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 | CAGATGCCACAGTCAACACA | GGCTCAGCTATGAAAGCAGAA | 9 | |
| NFKB | Nuclear Factor Kappa B | ACCCAAGGACATGGTGGTC | AGCCCCTTATACACGCCTCT | 47 | |
| SIRT2 | Sirtuin 2 | TTCAAGCCAACCATCTGTCA | GCTCCAGGGTATCTATGTTCT | 40 | |
| PGC1a | PPARG Coactivator 1 Alpha | AAACGATGACCCTCCTCACA | TTCTTTTTGGAGGTGCATTTG | 84 | |
| PARP1 | Poly(ADP-Ribose) Polymerase 1 | GACAGGAAAGACAACAGACAAATC | GGGGTGATGTGTTTGAACTTG | 7 | |
| TNFA | Tumor Necrosis Factor-Alpha | CAGCCTCTTCTCCTTCCTGAT | GCCAGAGGGCTGATTAGAGA | 40 | |
| p53 | Tumor Protein P53 | TAGTGTGGTGGTGCCCTATG | CACATGTAGTTGTAGTGGATGGT | 21 | |
| Cellular Significance | Gene | Full Gene Name | Changes in Expression Level | Function of the Encoded Protein | |
|---|---|---|---|---|---|
| KR | 7-deazaKR | ||||
| Enzymatic scavengers involved in the antioxidant defense | SOD | Superoxide Dismutase | ↑↑↑ | ↑↑ | Involved in the antioxidant defense against oxidative stress; SOD catalyzes the dismutation of superoxide anion to hydrogen peroxide. |
| CAT | Catalase | ↑↑ | ↑ | The key antioxidant enzyme catalyzing the decomposition of hydrogen peroxide into water and oxygen. | |
| GSS | Glutathione Synthetase | ↑ | ↑↑ | The important enzyme of cellular antioxidant defense; involved in the second step of biosynthesis of glutathione (GSH), one of the functions of which is to protect cells from oxidative damage by free radicals. | |
| Regulating factors related to oxidative stress | SESN1 | Sestrin 1 | ↑ | ↑↑↑ | Sestrins are induced by the p53 protein and play a role in the cellular response to DNA damage and oxidative stress. |
| SESN2 | Sestrin 2 | no change | ↑↑↑ | ||
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 | ↑ | ↑↑↑ | A transcription factor that controls the expression of antioxidant proteins involved in oxidative damage protection. | |
| NFKB | Nuclear Factor Kappa B | ↑↑ | ↑↑↑ | A transcription regulator that is activated by various intra- and extracellular stimuli, including oxidant free radicals. | |
| Cellular welfare indicators | PGC-1a | PPARG Coactivator 1 Alpha | ↓↓↓ | ↓↓↓ | A transcriptional coactivator of the genes involved in energy metabolism. It interacts with and regulates the activities of nuclear respiratory factors (NRFs). |
| SIRT2 | Sirtuin 2 | ↑↑ | ↑↑ | Involved in protection against various types of cellular stress related to oxidative stress (e.g., upregulates the expression of FOXO3 target gene, decreasing ROS level); involved in DNA repair. | |
| PARP1 | Poly(ADP-Ribose) Polymerase 1 | ↑ | ↑↑ | Involved in the regulation of the molecular events related to the recovery of cells from DNA damage. | |
| TNFA | Tumor Necrosis Factor-Alpha | ↑↑↑ | ↑ | Multifunctional proinflammatory cytokine, involved in the regulation of a wide spectrum of biological processes, including cell proliferation, differentiation, apoptosis, and lipid metabolism. | |
| p53 | Tumor Protein P53 | ↑↑ | ↑↑↑ | The encoded protein responds to diverse cellular stresses to regulate the expression of target genes, thereby inducing cell-cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Gurda-Woźna, D.; Pawelczak, P.; Krawczyk, P.; Giel-Pietraszuk, M.; Framski, G.; Ostrowski, T.; Wyszko, E. Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives. Antioxidants 2021, 10, 950. https://doi.org/10.3390/antiox10060950
Orlicka-Płocka M, Fedoruk-Wyszomirska A, Gurda-Woźna D, Pawelczak P, Krawczyk P, Giel-Pietraszuk M, Framski G, Ostrowski T, Wyszko E. Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives. Antioxidants. 2021; 10(6):950. https://doi.org/10.3390/antiox10060950
Chicago/Turabian StyleOrlicka-Płocka, Marta, Agnieszka Fedoruk-Wyszomirska, Dorota Gurda-Woźna, Paweł Pawelczak, Patrycja Krawczyk, Małgorzata Giel-Pietraszuk, Grzegorz Framski, Tomasz Ostrowski, and Eliza Wyszko. 2021. "Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives" Antioxidants 10, no. 6: 950. https://doi.org/10.3390/antiox10060950
APA StyleOrlicka-Płocka, M., Fedoruk-Wyszomirska, A., Gurda-Woźna, D., Pawelczak, P., Krawczyk, P., Giel-Pietraszuk, M., Framski, G., Ostrowski, T., & Wyszko, E. (2021). Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives. Antioxidants, 10(6), 950. https://doi.org/10.3390/antiox10060950