1. Introduction
Hydrogen peroxide (H
2O
2) is a major reactive oxygen species (ROS) that acts as an intracellular signaling molecule by selective oxidation of cysteines on proteins. In doing so, H
2O
2 regulates several biological activities, such as cell proliferation and differentiation, tissue repair, inflammation, the circadian rhythm, and aging [
1]. The major endogenous H
2O
2 sources are the mitochondrial respiratory chain and the NADPH oxidases (NOX), which release H
2O
2 in a regulated, localized manner [
2,
3,
4]. To ensure that H
2O
2 levels stay within the nanomolar range and it does not accumulate to levels where it may exert irreversible damage to biomolecules, cells also harbor H
2O
2 scavenging systems. These include catalases, glutathione peroxidases (Gpxs), and peroxiredoxins (Prdxs) [
1,
3,
5]. Among them, Prdxs are responsible for the majority of H
2O
2 reduction within cells as Prdxs are highly abundant (≈1% of total soluble protein content) and very efficient in removing peroxides (k ≈ 10
5–10
8 M
−1 s
−1) [
3,
5,
6,
7]. Prdxs scavenge H
2O
2 through the formation of a sulfenic acid (Cys-SOH) on the peroxidatic cysteine (Cys
P) with the subsequent formation of a disulfide bond between Cys
P and the resolving cysteine (Cys
R) [
8], and are recycled via the thioredoxin pathway (Trx-TrxR-NADPH) [
9]. This super-scavenging activity is due to the low pKa of the Cys
P and a conserved hydrogen bonding network which renders one of the oxygens of H
2O
2 more susceptible to nucleophilic attack by Cys
P [
10]. In the presence of such abundant and highly efficient H
2O
2 scavengers, for a long time it was unclear how even some low-abundant proteins, whose thiols typically have kinetic rate constants in the 10–10
2 M
−1 s
−1 range [
5], can get oxidized [
11,
12]. Accumulating evidence [
13,
14,
15,
16] suggests that the answer to this conundrum lies in the ability of the sulfenic acid formed on Cys
P of Prdxs to react with a thiol of a target protein, instead of Cys
R, or of the Cys
P–Cys
R disulfide to partake in thiol–disulfide exchange with nucleophilic thiols of target proteins. Both lead to the formation of a mixed disulfide with the target protein via which the oxidative equivalents are transferred in a process termed “redox-relay”. This mixed disulfide can then rearrange into an intramolecular disulfide on the target protein, which can affect the protein’s localization, conformation, interaction partners, or functionality [
17,
18,
19,
20,
21,
22,
23].
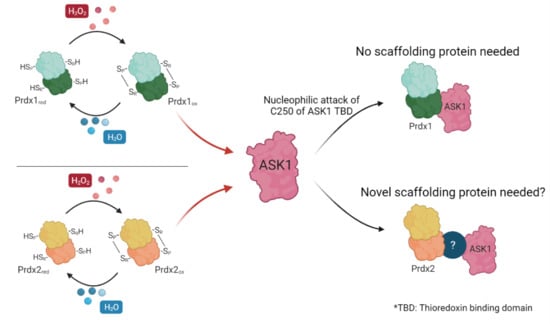
The first documented example of such redox-relays came from budding yeast, where the thiol peroxidase Orp1 was shown to specifically transfer oxidative equivalents to the transcription factor Yap1 through a direct protein–protein interaction (PPI) in the presence of H
2O
2. As a result of this oxidation by Orp1, Yap1 is retained in the nucleus launching an adaptive transcriptional program [
13]. Ten years after the discovery of the Orp1:Yap1 redox-relay, the first evidence of such redox-relays in the cytosol of mammalian cells was reported by Jarvis et al. [
14], who demonstrated that Prdx1 facilitates the oxidation of the apoptosis signal-regulating kinase 1 (ASK1). ASK1 is a mitogen-activated protein kinase kinase kinase (MAP3K) which phosphorylates downstream kinases, eventually leading to the activation of the c-Jun N-terminal kinase (JNK) and p38 MAP kinase pathways [
24,
25,
26]. Activation of these pathways, in turn, can result in the upregulation of important targets such as AP-1 (c-Jun family member), NFκB, and STAT1, all of which are implicated in cancer [
27,
28,
29] (
Figure 1). To exert its kinase activity, ASK1 needs to be oxidized to disulfide-bonded multimers, with Cys250 playing a critical role [
30,
31]. ASK1 multimers are then reduced by Trx1 [
30]. In addition, reduced Trx1 regulates ASK1 activity by non-covalently binding to the so-called Trx-binding domain (ASK1-TBD) in the vicinity of Cys250 [
25,
32]. The inhibitory effect of this binding can be explained by the fact that it prevents ASK1 multimerization or the formation of other potential disulfide bonds that could alter its tertiary or quaternary structure and lead to activation with basal levels of H
2O
2 [
31]. Upon exposure to an H
2O
2 signal (which can differ from basal levels by up to 50-fold, reaching 500–700 nM [
33]), Trx1 becomes oxidized and dissociates from ASK1 (
Figure 1), thereby allowing ASK1 to form disulfide-bonded multimers, become auto-phosphorylated, and unleash the ASK1-p38/JNK cascade [
24,
25,
26]. The discovery of the Prdx1:ASK1 redox-relay, therefore, fit well with this model of H
2O
2-mediated ASK1 activation, with oxidized Prdx1 forming a putative mixed disulfide with Cys250 of ASK1 after the dissociation of Trx1 from ASK1-TBD.
Though the paper by Jarvis et al. (2012) [
14] has been widely cited as the first Prdx-mediated redox-relay in the cytosol of mammalian cells, evidence for this relay is based on only a few experiments. The first is indirect: an attenuation of ASK1 oxidation and p38 phosphorylation was seen upon Prdx1 knock down. The second piece of evidence comes from the comparison of reducing and non-reducing immunoblots against ASK1, where mixed disulfides corresponding in size to ASK1:Prdx1 intermediates could be observed. A co-immunoprecipitation experiment on cell lysates then revealed that ASK1 co-precipitates with Prdx1, and that this mixed disulfide can be reduced by DTT [
14]. Even though the authors took care in preventing artificial oxidation upon cell lysis by carrying out the experiment in the presence of high amounts (100 mM) of NEM, it was reported that thiol blocking under these conditions is not fully efficient [
15,
34]. To our knowledge, no further attempts of proving the Prdx1:ASK1 interaction by other means, including those that do not require cell lysis, have been undertaken.
In stark contrast, the second example of cytosolic Prdx-mediated redox-relays in mammalian cells, between Prdx2 and the transcription factor STAT3 (signal transducer and activator of transcription 3), received much more attention [
15,
35]. Indeed, the Prdx2:STAT3 redox has been confirmed in several experimental settings on cell lysates (co-immunoprecipitation), as well as in both fixed (proximity ligation assay) and live cells (bimolecular fluorescence complementation (BiFC) and Förster resonance energy transfer (FRET)). Together, these results gave more insight into the Prdx2:STAT3 interaction, revealing that Prdx2 and STAT3 already interact prior to and independent of H
2O
2, and are localized to membrane-associated microdomains, as well as this interaction being specific to Prdx2 over Prdx1. Interestingly, the only type of experiment where the Prdx2:STAT3 interaction was not observed was in vitro on recombinant proteins [
35]. This discrepancy between in vitro and cellular experiments then led to the discovery that the transfer of oxidative equivalents from Prdx2 to STAT3 is facilitated by annexin A2 (AnxA2) [
35], similar to how Ybp1 mediates the Orp1:Yap1 interaction in
S. cerevisiae [
36].
Inspired by the exciting insights into mammalian cytosolic Prdx redox-relays yielded by Prdx2:STAT3 interaction studies, in this study, we revisit the Prdx1:ASK1 redox-relay. We utilize BiFC as an approach for studying PPI in cells and bio-layer interferometry (BLI) as an in vitro PPI method to validate and gain more insight into the interaction reported a decade ago. Our results show that, unlike Prdx2 and STAT3, Prdx1 and ASK1 only interact upon elevation of cytosolic H2O2 levels above exogenous ones, and independently of a facilitator protein. Further, we found that Prdx2 can also bind to ASK1 in cells even after knocking out AnxA2, but not in vitro, suggesting the need for a facilitator protein that is not AnxA2.
2. Materials and Methods
2.1. Ab Initio Modeling by I-TASSER and Docking Simulations with HADDOCK
The I-TASSER (Iterative Threading Assembly Refinement) (Zhang Lab) webserver (
https://zhanglab.ccmb.med.umich.edu/I-TASSER/, accessed in 30 November 2020) was used for the 3D ab initio modeling of the eight different Prdx1- and ASK1-mLumin fusion proteins [
37]. The N- (LN) or C-terminal (LC) half of mLumin was fused to either the N- or C-terminus of Prdx1 and ASK1 separated by a linker. The linker for the LN fragment is: Gly-Ser-Tyr-Pro-Tyr-Asp-Val-Pro-Asp-Tyr-Ala-Gly-Thr-Gly-Gly-Ser-Lys-Ser-Thr; and for the LC is: Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu-Gly-Thr-Gly-Gly-Ser-Lys-Ser. The models obtained were superimposed with the human ASK1 kinase domain (PDB 2CLQ) [
38] and the human Prdx1 C83S mutant (PDB 4XCS) [
39] using PyMol (Version 2.4.1, Schrödinger, Inc., New York, USA) [
40], and models where the secondary structure was maintained (low r.m.s.d.) were selected. These models were then submitted to the High Ambiguity Driven Biomolecular Docking (HADDOCK) (Bonvin Lab) [
41] webserver (
https://wenmr.science.uu.nl, accessed in 30 November 2020) to identify the most stable docking between Prdx1 and ASK1 fused to complementary mLumin fragments. The defined criteria for running the docking were: ‘All’ chains were defined to be involved in the interaction; the peroxidatic Cys52 of Prdx1 [
42] and the Cys located in the Trx binding domain (TBD) (120, 185, 200, 206, 225, 226, and 250) of ASK1 [
32] were specified as active residues; and residues surrounding the active residues were automatically defined as passive.
2.2. Cell Lines, Antibodies, Chemicals, and Plasticware
The HEK293 MSR (Griptite
TM, Thermo Fisher Scientific. Waltham, WA, USA) and annexin A2 knock-out (AnxA2 KO) HEK293 MSR cells (gift from the lab of Tobias Dick, German Cancer Research Center, Heidelberg, Germany) [
35] were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Carlsbad, CA, USA) and 50 units/mL penicillin and streptomycin (P/S) (Life Technologies). Cell lines were routinely checked for the absence of mycoplasma using the PCR Mycoplasma Test Kit (PromoCell, Cat.PK-CA91-1024, Heidelberg, Germany). Plasticware for cell culture was from Avantor (Radnor, PA, USA).
Antibodies used in this study were rabbit anti-Prdx1 (D5G12) (8499, Cell Signaling Technology®, Danvers, MA, USA), mouse anti-Prdx2 (WH0007001M1, Sigma-Aldrich, St. Louis, MO, USA), mouse anti-ASK1 (MA5-15861, ThermoFisher Scientific), mouse anti-STAT3 (9139S, Cell Signaling Technology®, Danvers, Massachusetts, USA), mouse anti-His (AD1.1.10, Bio-Rad Laboratories, Richmond, VA, USA), rabbit anti-β-tubulin (2146S, Cell Signaling Technology®, Danvers, MA, USA), and mouse anti-β-actin (A2228, Sigma-Aldrich, St. Louis, MO, USA). All antibodies were used at a dilution of 1:1000 in Tris-buffered saline with 0.1% Tween 20 (TBS-T), except anti-Prdx2 which was used at a dilution of 1:5000 in TBS-T. The secondary antibodies for Western blot used in this study were anti-mouse IgG (A3562, Sigma-Aldrich, St. Louis, MO, USA) and anti-rabbit IgG (A8025, Sigma-Aldrich, St. Louis, MO, USA). All secondary antibodies were alkaline phosphatase conjugated and used at a dilution of 1:10000, also in TBS-T. The protein ladder used in SDS-PAGE was PageRuler Prestained protein ladder (Thermo Fisher Scientific, Waltham, WA, USA).
All chemicals were purchased from Merck (Sigma-Aldrich, St. Louis, MO, USA) unless stated otherwise.
2.3. Cloning of Prdx1, Prdx2, ASK1, and STAT3 DNA Constructs
The mammalian cell expression vector pcDNA3.1(−) harboring either the N- (LN) or C-terminal (LC) half of mLumin with the respective linker and the pcDNA3.1(−)-STAT3 LN, pcDNA3.1(−)-Prdx2 WT-LC, and pMAX-GFP constructs were kindly provided by the lab of Tobias Dick (German Cancer Research Center, Heidelberg, Germany). Prdx1 and ASK1 genes (codon-optimized for mammalian cells) synthesized at Genscript (Piscataway, NJ, USA) were cloned into the pcDNA3.1(−)-mLumin vectors using the NEBuilder
® HiFi DNA Assembly Master Mix (NEB Cat. E2621) (New England Biolabs, Ipswich, MA, USA), in the orientation predicted by the in silico modeling and docking simulations. Primers for the HiFi DNA Assembly were designed using Snapgene (version 5.2.2. GSL Biotech. San Diego, CA, USA). Peroxidatic Cys (C52A) and resolving Cys (C173A) mutants of Prdx1 were generated by site-directed mutagenesis using the Pfu DNA polymerase (Agilent Technologies, Santa Clara, CA, USA). Primers for mutagenesis were designed by PrimerX (
https://www.bioinformatics.org/primerx/, accessed in 30 May 2020). All primers are listed in (
Supplementary Table S1). Plasmids were purified using the Plasmid Plus Midi kit (Qiagen, Hilden, Germany).
Prdx1 and Prdx2 WT constructs for recombinant protein expression in
E. coli (pET17-Prdx1 and pET17-Prdx2), which have already been used in Prdx structural studies [
43,
44], were kindly provided by the lab of Todd Lowther (Wake Forest School of Medicine, Winston-Salem, NC, USA). The ASK1 thioredoxin-binding domain (ASK1-TBD) (codon-optimized for
E. coli) was expressed using a pEH vector harboring an N-terminal 6x-His tag along with a maltose binding protein (MBP) and tobacco etch virus (TEV) cleavage site. This construct was purchased from the VIB protein core (Gent University, Gent, Belgium).
All constructs were confirmed by Sanger sequencing.
2.4. Transfection of Cell Lines
A total of 2 × 105 HEK293 MSR or AnxA2 KO HEK293 MSR cells were seeded per well of a 12-well plate. The next day, cells were co-transfected with two plasmids (either mLumin-ASK1 or mLumin-STAT3 with mLumin-Prdx (Prdx1 WT, Prdx1 C52A, Prdx1 C173A, or Prdx2 WT)) in equimolar ratio. Transfection was performed using the Lipofectamine 3000 kit (Thermo Fisher Scientific, Waltham, WA, USA) following the protocol provided by the manufacturer. A GFP-encoding plasmid was also co-transfected for normalization purposes in an equimolar ratio with the two plasmids above.
2.5. Bimolecular Fluorescent Complementation Assay and Image Analysis
24 h after transfection, cells were treated with either a mixture of xanthine/xanthine oxidase (8 µM of xanthine and 1 mU/mL of xanthine oxidase) (X/XO), 0.8 µM auranofin for 18 h, or a bolus of 100 µM H2O2 for 30 min. All the oxidants were prepared in DMEM supplemented with 10% FBS and 50 units/mL P/S and this oxidant-containing DMEM then replaced the DMEM cells were growing in (total volume: 1 mL). As each well contained 0.4 × 106 cells, we estimate that in case of 100 µM H2O2, each cell was exposed to 250 femtomoles of H2O2. The amount of H2O2 each cell experienced upon treatment with X/XO and auranofin is more difficult to estimate.
18 h after treatment with X/XO or auranofin, or after 30 min of H2O2 treatment, mLumin fluorescence and GFP fluorescence images were captured using a fluorescence microscope (Leica DMi8, Wetzlar, Germany), using the 10× (506406) objective. Samples were excited with the 561 nm (for mLumin) and 488 nm (for GFP) laser lines and fluorescence was detected with a 600–680 nm filter for mLumin, and 500–580 nm for GFP.
All images were captured in RAW format and the mLumin and GFP fluorescence-integrated intensities (i.e., the sum of all the pixel values with intensities in a certain threshold selected to avoid saturation) were determined using ImageJ (
https://imagej.nih.gov/ij/, accessed in 30 July 2020). The final readout (mLumin intensity normalized to GFP intensity) was obtained by dividing the integrated intensity of mLumin by that of GFP in the same threshold range.
2.6. Validation of Bimolecular Fluorescent Complementation Construct Expression
A total of 4.8 × 105 HEK293 MSR or AnxA2 KO HEK293 MSR cells were seeded per well of a 6-well plate. After 24 h, cells were co-transfected with two plasmids encoding the mLumin constructs as described above. Co-transfected cells were harvested after 48 h, pelleted by centrifugation at 4500× g for 6 min at 4 °C, resuspended in lysis buffer (20 mM Tris/HCl, pH 7.4, cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland), 0.1 mM EDTA, 0.5 mM AEBSF, and 0.5 mM MgCl2), and rotated for 30 min at 4 °C, and the lysate was clarified by centrifuging at 16,000× g for 20 min at 4 °C. Protein concentration was determined using the Bio-Rad Protein assay (Cat. 500-0006, Bio-Rad Laboratories, Richmond, VA, USA).
The protein samples were separated by SDS-PAGE and transferred to polyvinyl difluoride (PVDF) membranes (Immobilon-P, Millipore, Burlington, MA, USA) using the Trans-Blot® TurboTM transfer system (Bio-Rad Laboratories, Richmond, VA, USA). Membranes were probed with appropriate antibodies and visualized by the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate p-nitroblue tetrazolium chloride (BCIP/NBT) (Abcam, Cambridge, UK).
2.7. Purification of Recombinant Proteins
All pellets of recombinant proteins in this paper were prepared following the same protocol unless stated otherwise. All proteins were expressed in the BL21 E. coli strain (New England Biolabs, Ipswich, MA, USA).
2.8. Purification of Recombinant Wild-Type Peroxiredoxin-1 (Prdx1 WT) and Prdx1 Mutants (Prdx1 C52A and Prdx1 C173A)
For expression of the Prdx1 WT and mutant constructs, 1 L LB media with ampicillin (100 μg/mL) was inoculated with a 100-fold dilution of an overnight pre-culture and grown at 37 °C with shaking at 120 rpm until the exponential growth phase was reached (OD600 = 0.4−0.6), then cooled to 16 °C, induced with 0.4 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG), and further grown overnight at 16 °C with shaking at 120 rpm.
The purification of Prdx1 WT and Prdx1 mutants (C52A and C173A) were performed following the same protocol. Cells were pelleted by centrifugation at 6150 × g for 15 min at 4 °C and resuspended in lysis buffer (50 mM HEPES/NaOH, pH 7.4, 250 mM NaCl, 0.1 mg/mL AEBSF, 1 µg/mL Leupeptin, 50 µg/mL DNaseI, and 20 mM MgCl2), lysed in a cell cracker at 20 kPsi at 4 °C, and the lysate was clarified by centrifuging at 39,846 × g for 30 min at 4 °C. Then, 20% of ammonium sulfate was added to the collected supernatant and centrifuged again at 39,846 × g for 30 min at 4 °C to remove the precipitated proteins. After that, the supernatant was passed through a 0.45 µM filter and loaded onto a Phenyl Sepharose® column (Cytiva, Marlborough, MA, USA) equilibrated with binding buffer (50 mM HEPES/NaOH, pH 7.4, 0.1 mM ethylenediaminetetraacetic acid disodium salt (EDTA), 20% ammonium sulfate, 2 mM dithiothreitol (DTT)). The unbound material was removed by washing the resin with 10 column volumes of binding buffer, after which the bound protein was gradient eluted with 10 column volumes of binding buffer without ammonium sulfate. The fractions containing Prdx1 (determined by running on SDS-PAGE gel in both non-reducing and reducing conditions (50 mM DTT)) were collected and dialyzed overnight with 4 buffer changes in the dialysis buffer (50 mM HEPES/NaOH, pH 7.4, 10 mM NaCl, 0.1 mM EDTA, 2 mM DTT) at 4 °C. The dialyzed protein sample was subsequently loaded onto a cation exchange SP Sepharose® Fast Flow column (Cytiva, Marlborough, MA, USA). After the unbound proteins were eliminated by washing with 10 column volumes of the binding buffer with the same composition as the dialysis buffer, the proteins were eluted with a 10 column volumes gradient of elution buffer (binding buffer with 1 M NaCl). The fractions containing Prdx1 were collected and concentrated using a Vivaspin with a 20 kDa cut-off (Sartorius, Göttingen, Germany), and injected onto a size-exclusion SuperdexTM75 16/600 column (Cytiva, Marlborough, MA, USA) equilibrated with 50 mM HEPES/NaOH, pH 7.4, 150 mM NaCl, 0.1 mM EDTA, 5 mM Tris(2-carboxyethyl)phosphine (TCEP)). Prdx1-containing fractions were collected, concentrated, and the protein concentration was determined spectroscopically using an extinction coefficient of 18,450 M−1 cm−1. The protein sample was flash-frozen in liquid nitrogen and stored at −80 °C.
2.9. Purification of Recombinant Wild-Type Prdx2 (Prdx2 WT)
Cultures of cells expressing Prdx2 WT were grown as described above for Prdx1. Cells were pelleted by centrifugation at 6150× g for 15 min at 4 °C and resuspended in lysis buffer (20 mM HEPES/NaOH, pH 7.4, 100 mM NaCl, 0.1 mM EDTA), lysed in a cell cracker at 20 kPsi at 4 °C, and the lysate was clarified by centrifuging at 39,846× g for 30 min at 4 °C. Next, 20% of ammonium sulfate was added to the clarified lysate, followed by centrifugation at 39,846× g for 30 min at 4 °C. Then, the collected supernatant was passed through a 0.45 µM filter and loaded onto a Phenyl Sepharose® column (Cytiva, Marlborough, MA, USA) equilibrated with binding buffer (20 mM HEPES/NaOH, pH 6.5, 1 mM EDTA, 20% ammonium sulfate). The unbound material was removed by washing the resin with 10 column volumes of binding buffer, after which the bound protein was gradient eluted with 10 column volumes of elution buffer without ammonium sulfate. The fractions containing Prdx2 (determined by running on SDS-PAGE gel in both non-reducing and reducing conditions (50 mM DTT)) were collected and dialyzed overnight with 4 buffer changes in the dialysis buffer (20 mM Tris/HCl, pH 7.9) at 4 °C. Next, the dialyzed protein sample was loaded onto an anion exchange Q Sepharose® Fast Flow column (Cytiva, Marlborough, MA, USA). After the unbound proteins were eliminated by washing with 10 column volumes of the binding buffer with the same composition as the dialysis buffer, the proteins were gradient eluted with 10 column volumes of elution buffer (binding buffer with 0.5 M NaCl). The fractions containing Prdx2 were collected and dialyzed overnight to 20 mM HEPES/NaOH, pH 7.5 with 4 buffer changes at 4 °C. The dialyzed protein sample was collected and concentrated using a Vivaspin with a 20 kDa cut-off (Sartorius, Göttingen, Germany), and injected onto a size-exclusion SuperdexTM200 16/600 column (Cytiva, Marlborough, MA, USA) equilibrated with 20 mM HEPES/NaOH, pH 7.5. Prdx2-containing fractions were collected, concentrated, and the protein concentration was determined spectroscopically using an extinction coefficient of 21,555 M−1 cm−1. The protein was flash-frozen in liquid nitrogen and stored at −80 °C.
2.10. Purification of the Recombinant Thioredoxin-Binding-Domain of ASK1 (ASK1-TBD)
The pellet of cells expressing 6xHis-MBP-ASK1-TBD was lysed in 50 mM HEPES/NaOH, pH 7.5, 250 mM NaCl, 0.1 mg/mL AEBSF, 1 µg/mL Leupeptin, 50 µg/mL DNaseI, and 20 mM MgCl2 and clarified the same way as Prdx1 and Prdx2. The supernatant was passed through a 0.45 µM filter and loaded onto a Ni2+-Sepharose® 6 Fast Flow column (Cytiva, Marlborough, MA, USA) equilibrated with binding buffer containing 50 mM HEPES/NaOH, pH 7.5, 150 mM NaCl, and 20 mM imidazole. The unbound proteins were eliminated by washing the resin with 10 column volumes of binding buffer. The MBP-ASK1-TBD protein was eluted with a linear 10 column volume gradient with elution buffer (50 mM HEPES/NaOH, pH 7.5, 150 mM NaCl, 500 mM imidazole). The fractions containing MBP-ASK1-TBD (determined by SDS-PAGE) were pooled and dialyzed overnight to 50 mM HEPES/NaOH, pH 7.5, 150 mM NaCl, 0.1 mM EDTA, 5 mM DTT with 4 buffer changes at 4 °C. The sample was concentrated using a 20 kDa cut-off Vivaspin concentrator (Sartorius, Göttingen, Germany) and injected onto a size-exclusion SuperdexTM75 16/600 column (Cytiva, Marlborough, MA, USA) equilibrated with 50 mM HEPES/NaOH, pH 7.5, 150 mM NaCl, 0.1 mM EDTA, 5 mM TCEP. Fractions containing MBP-ASK1-TBD were collected and concentrated using the same Vivaspin 20 kDa cut-off concentrator. The protein concentration was determined spectroscopically using an extinction coefficient of 81,250 M−1 cm−1. The protein was flash-frozen in liquid nitrogen and stored at −80 °C.
After purification, all recombinant proteins were validated by Western blot. The protein samples were separated by SDS-PAGE and transferred to polyvinyl difluoride (PVDF) membranes (Immobilon-P, Millipore) using Trans-Blot® TurboTM transfer system (Bio-Rad Laboratories, Richmond, VA, USA). Membranes were probed with appropriate antibodies and visualized by the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate p-nitroblue tetrazolium chloride (BCIP/NBT) (Abcam, Cambridge, UK).
2.11. Ferrous Oxidation-Xylenol Orange (FOX) Assay
Prdx1 WT, Prdx1 C52A, and Prdx1 C173A were reduced with 50 mM DTT for 30 min at room temperature. DTT was removed using a Hitrap® desalting column (Cytiva, Marlborough, MA, USA) equilibrated with 50 mM HEPES/NaOH, pH 7.4, 100 mM NaCl. The protein concentration was determined spectroscopically using an extinction coefficient of 18,450 M−1 cm−1 for Prdx1.
The FOX assay was performed based on the protocol described by Nelson et al. (2011) [
45]. Briefly, the FOX reagent was prepared by mixing 1 part of FOX A with 100 parts of FOX B. FOX A consists of 25 mM ammonium ferrous sulfate in 2.5 M sulfuric acid (H
2SO
4), and FOX B consists of 100 mM sorbitol and 125 µM xylenol orange. Both FOX A and FOX B solutions were prepared in water.
Prior to the assay, the H2O2 stock concentration was determined by measuring the absorbance at 240 nm (ε240 = 43.6 M−1 cm−1). To quantify the H2O2 concentrations, a calibration curve of increasing concentrations of H2O2 (0 µM to 250 µM) was created. For this purpose, 10 µL of each H2O2 solution was mixed with 490 µL FOX reagent, vortexed, and incubated in the dark for 30 min. 200 µL of the colorimetric solution was then transferred to the microtiter clear 96-well plate (Thermo Fisher Scientific) and the absorbance was measured at 560 nm using the Spectramax 340PC microplate reader (Molecular Devices, San Jose, CA, USA). Absorbance was plotted vs. concentration. The data were fitted with a linear regression and the R2 was determined.
Protein samples were mixed with 200 µM H2O2 and 1 µM of DTT at a final protein concentration of 2 µM. At time-points 1, 2, 5, 7, and 10 min, 10 µL of the protein-H2O2 sample was mixed with 490 µL FOX reagent, vortexed, and incubated in the dark for 30 min. Then, 200 µL of the colorimetric solution were transferred to the microtiter clear 96-well plate and absorbance was measured at 560 nm. The concentration of the remaining H2O2 in the sample was interpolated using the regression equation from the calibration curve of H2O2.
2.12. Horseradish Peroxidase (HRP) Assay for Determination of kSOH
The second-order rate constant of the pre-reduced Prdx recombinant proteins (WT and C173A) was determined to confirm the expected active function. For this, the HRP assay was used to monitor the Prdx’s ability to compete with HRP in the reduction of H
2O
2, as described [
45]. Briefly, in 96-well UV plates, in a total volume of 150 µL, six to eight different protein concentrations of purified Prdx (2–30 µM) were mixed with HRP and H
2O
2 to obtain 15 µM HRP (Sigma-Aldrich, St. Louis, MO, USA) and 3 µM H
2O
2 final concentrations and the absorbance was measured before and within 60 s of the start of the reaction. HRP forms compound I upon reaction with H
2O
2, which can be monitored as a decrease in absorbance at 403 nm [
46]. When Prdx outcompetes HRP for the H
2O
2, the decrease in absorbance is less because less compound 1 is formed. Thus, data collected at different Prdx concentrations can be transformed to k
SOH by plotting the change in absorbance against Prdx concentration. This relationship is represented by this equation:
Experiments were done in triplicate for each Prdx concentration and repeated with fresh aliquots of enzymes.
2.13. Bilayer Interferometry (BLI)
For the BLI assay on Octet
Red 96 (ForteBio, Fremont, CA, USA), proteins were reduced with 20 mM DTT for 30 min at room temperature. DTT was removed using a Hitrap
® desalting column (Cytiva, Marlborough, MA, USA) equilibrated in 25 mM Tris/HCl, pH 7.4, 25 mM NaCl. To block all free Cys, Prdxs, MBP-ASK1-TBD, and MBP were treated with 20 mM iodoacetamide (IAM) at room temperature for 30 min, and excess IAM was removed on Bio-spin columns (Bio-Rad Laboratories, Richmond, VA, USA). Next, the number of free thiols was determined using Ellman’s reagent (5,5-dithio-bis-(2-nitrobenzoic acid)) (DTNB assay) to confirm that all potential thiol groups were blocked [
47].
The protein MBP was used as a reference (negative control) to eliminate the binding possibility of Prdx1 WT to the MBP part of the MBP-ASK1 fusion protein, and Prdx2 WT was used to determine the selectivity of the Prdx1:MBP-ASK1-TBD interaction. All proteins were prepared in 25 mM Tris/HCl, pH 7.4, 25 mM NaCl, 1% bovine serum albumin (BSA), 0.05% Tween 20, and 0.01 mM maltose. His-tagged MBP-ASK1-TBD and His-tagged MBP were loaded onto the Ni
2+-NTA sensors (ForteBio, Fremont, CA, USA) at a concentration of 0.45 µM and 0.15 µM, respectively. The concentration of analytes (Prdx1 WT, Prdx1 C52A, Prdx1 C173A, and Prdx2 WT) was fixed at 1 µM. The assay was done in the presence of 10 µM DTT for the reducing condition and 10 µM H
2O
2 for the oxidizing condition. Data were obtained with the Data Acquisition 9.0 (ForteBio, Fremont, CA, USA) software of the instrument. To calculate association (
kon) and dissociation rate (
koff) constants, different concentrations of the analyte (Prdx1s) were utilized, and the data were analyzed by the Data Analysis 9.0 software (ForteBio, Fremont, CA, USA). In this software, the association curves were fitted through the equation:
The dissociation curves were fitted through the following equations:
where
y is the BLI signal (in nm), indicating the level of binding as nm shift, while
y0 represents the nm shift at the beginning of the dissociation phase,
t is the time (s), and
t0 is the time at the beginning of the dissociation phase. [
Analyte] is the given concentration of Prdx1 or its variants, and
Rmax is the fitted maximum binding of the analyte to a given immobilized ligand on the biosensor surface.
2.14. Circular Dichroism (CD)
The purified Prdxs (WT, C52A, and C173A) were pre-reduced with 20 mM DTT at room temperature for 30 min, and excess DTT was subsequently removed using a Hitrap
® desalting column (Cytiva, Marlborough, MA, USA) equilibrated with 10 mM potassium phosphate buffer pH 7.4, 100 mM potassium fluoride (KF). The protein concentrations were determined spectrophotometrically, using an extinction coefficient of 18,450 M
−1 cm
−1. Then, the Prdxs were treated with H
2O
2 at 1/10 molar ratio of Prdx/H
2O
2 for 30 min at room temperature. Excess H
2O
2 was removed using Bio-spin columns (Bio-Rad Laboratories, Richmond, VA, USA) following the user manual and then Prdxs were concentrated to 0.1 mg/mL and 0.05 mg/mL for the reduced and oxidized samples, respectively. The CD spectrum (190–260 nm) was recorded using the CD Spectrometer J-175 (Jasco, Tokyo, Japan). The molar ellipticity [
θ] was calculated using the equation:
where
θ is the ellipticity in degree,
M is the molecular mass,
C is the concentration of protein in mg/mL,
l is the pathlength in cm, and
n is the number of residues of the protein.
2.15. Data Analysis
Graphpad Prism (GraphPadSoftware, version 9.1.0. San Diego, CA, USA) was used for statistical analysis of the data. The statistical method used was two-way ANOVA and Dunnett’s multiple comparison test. The data are displayed as mean ± SD. A minimum significant level of p ≤ 0.05 was set.
4. Discussion
We decided to bring the first reported mammalian Prdx redox-relay, Prdx1:ASK1, back into the spotlight. Ever since co-immunoprecipitation experiments on cell lysates showed a putative mixed disulfide between Prdx1 and ASK1 [
14], there have been no published attempts to gain more mechanistic insights into this interaction or to at least validate it in intact cells.
Here, we confirmed the Prdx1:ASK1 interaction with an in-cell approach not requiring cell lysis, BiFC, as well as with in vitro experiments using recombinant proteins. We also discovered that Prdx1 and ASK1 interact in the absence of a scaffolding protein. Even though it has been reported that the central regulatory region of ASK1 can serve as a recruitment platform for ASK1 substrates [
52], our in vitro results clearly show that the TBD domain alone is sufficient for the Prdx1:ASK1 interaction, thus ruling out the requirement of another domain of ASK1 acting as a scaffolding protein. Further, we revealed that the Prdx1:ASK1 interaction only occurs upon H
2O
2 induction and depends on the peroxidatic cysteine (C52) of Prdx1. Most of these aspects are in total contrast to the well-characterized Prdx2:STAT3 interaction, where Prdx2 and STAT3 are known to associate even prior to induction with H
2O
2 and require AnxA2 for a productive oxidative transfer [
35].
There are several possible reasons for why Prdx2 and STAT3 require a scaffolding protein to interact, whereas Prdx1 and ASK1 do not, some of which could be related to the precise role of a scaffolding protein. On the whole, it is difficult to generalize on the function of scaffolding proteins for redox-relays, as only two such proteins have been reported—apart from the aforementioned AnxA2 [
35], Ybp1 mediates the Orp1:Yap1 relay in yeast [
36]. One potential role could be driven by kinetic arguments. Once the sulfenic acid is formed on the peroxidatic cysteine (C
P-OH) of Prdx1 or Prdx2, it could succumb to two fates: (i) it could be attacked by the thiol of the interactor forming a mixed disulfide bond, or (ii) it could condense with the resolving cysteine of Prdx to form an intersubunit C
P-C
R, after which the oxidative equivalents would be transferred to the interactor by thiol–disulfide exchange. Mathematical modeling of the Prdx2:STAT3 redox-relay showed that both mechanisms fit the experimental data well [
53]. However, the oxidized form of Prdx (both the sulfenic acid and the C
P-C
R intersubunit disulfide) is also competing with the mM concentrations of glutathione in the cytosol (reaction rate 500 M
−1 s
−1) [
54] or with thioredoxin (2.1 × 10
6 M
−1 s
−1) [
55]. Both reactions could outcompete the reaction with the thiol of the interactor, which is estimated to be in the 10
2 M
−1 s
−1 range for STAT3 [
53]. With this in mind, the role of the scaffolding protein would consist in shielding the Prdx C
P-OH or the intersubunit C
P-C
R disulfide from the cellular reducing systems, as suggested for Ybp1 [
36]. The rate constants of the condensation step in which the Prdx C
R nucleophilically attacks the C
P-OH differ by almost two orders of magnitude for Prdx1 and Prdx2 (11 s
−1 and 0.2 s
−1, respectively) [
56]. Hence, for Prdx2, there is an increased chance of this more long-lived C
P-OH to be attacked by glutathione compared to Prdx1 C
P-OH, which argues for an increased need of a scaffolding protein for Prdx2. One other unexplored possibility is post-translational modifications that could be extending C
P-OH lifetime through structural steric changes. Perhaps the different ways (i.e., PTM versus scaffolding protein) are the means to specificity in binding partner and binding process.
A scaffolding protein could also ensure that the interacting cysteines of Prdx and its interactor align properly for an efficient oxidative transfer, as observed for Orp1:Ybp1:Yap1 [
36]. Both STAT3 and ASK1 harbor multiple cysteine residues, but the difference may lie in the cellular location of the interacting partners. While the Prdx2:STAT3 interaction takes place at the membranes [
35], where the viscosity may render the rearrangements of the interacting proteins to obtain a proper alignment difficult, the Prdx1:ASK1 interaction is expected to take place in the cytosol [
57], which has a viscosity like that of pure water. Despite the molecular crowding of the cytosol, which slows down the diffusion of molecules about four-fold compared to water, the diffusion of macromolecules in membranes is still substantially slower [
58]. Interestingly, Orp1 and Yap1, which require Ybp1 to interact, or at least one of their pools, are also membrane-bound [
13]. Localization could also explain the need for Prdx2 and STAT3 to pre-assemble—due to the increased viscosity at the plasma membrane, the trafficking of the proteins is expected to be hindered; pre-assembly would significantly reduce the time required for the interaction to occur. Indeed, in vitro experiments have shown a clear dependence of the rates of catalyzed reactions and tertiary complex formation on viscosity [
59].
As mentioned above, as there have only been two peroxidase redox-relay complexes featuring a scaffolding protein characterized, all reasons on why the Prdx2:STAT3 redox-relay requires a scaffolding protein but the Prdx1:ASK1 relay does not will remain speculative. As such, the function of the Prdx interactor could also dictate the need for a scaffolding protein. Both Yap1 and STAT3 are transcription factors, whereas ASK1 is a kinase. Another kinase, MST1, was found to interact with Prdx1 in vitro, as assessed through a kinase assay, providing one more example of a Prdx:kinase interaction that occurs without a scaffolding protein. However, it is not clear whether Prdx1 and MST form a redox-relay [
60]. Whether these differences between ASK1 and STAT3 oxidation would hold true for other kinases and transcription factors, the physiological meaning of this, and where proteins with other functionalities that form redox-relays with Prdxs lie is a subject of future studies.
Our experiments with the peroxidatic (C52A) and resolving (C173A) mutants of Prdx1 resulted in in vitro vs. in-cell differences. This was especially striking for the peroxidatic cysteine mutant, where the interaction was completely abolished in vitro yet could still occur in cells. This demonstrates that the peroxidatic cysteine is essential for the interaction with ASK1 to occur, yet there are mechanisms in cells, most likely driven by endogenous Prdx1 and other proteins (to be discussed in detail below), that still enable the Prdx1 C52A:ASK1 association. It remains to be clarified if these alternative mechanisms result in true, productive redox-relays or if they are more general protein–protein interactions. Of note, the redox-inactive mutant of Prdx2 could still bind STAT3 and compete with wild type Prdx2 [
15].
As for the resolving cysteine mutant, while in cells it was indistinguishable from Prdx1 WT, a difference of less than one order of magnitude was observed in vitro. The reason behind this is likely to be purely technical: BiFC simply cannot capture small differences in reaction rates. The slightly slower k
on of the resolving cysteine mutant could be a consequence of minor local structural changes by which the efficiency of reactivity of C
P is partially being compromised, as reported recently for Prdx2 [
51]. The reaction of the resolving cysteine Prdx1 mutant with urate hydroperoxide was also lower than for the WT [
61]. The difference in reaction rates with ASK1 between the Prdx1 WT and resolving cysteine mutant that we observed hints to a Prdx1:ASK1 redox-relay that is preferentially formed by thiol–disulfide exchange (i.e., attack of ASK1 Cys250 on the Prdx1 C
P-C
R inter-subunit disulfide bond) rather than on the Prdx1 C
P-OH sulfenic acid. This would also be in line with the reaction rate of C
P-OH condensation with C
R (11 s
−1), which is two orders of magnitude faster than for Prdx2. Nevertheless, our in vitro results also clearly indicate that the procession through the C
P-OH can still occur, as otherwise we would not have observed an association of the Prdx1 C173A mutant with ASK1.
We also found that, unlike the Prdx2:STAT3 interaction, which is very specific for the peroxidase isoform [
35], Prdx2 can also interact with ASK1, but only in cells and to a lesser extent than Prdx1. This hints at the requirement of a scaffolding protein that is not AnxA2, perhaps quite expectedly, as ASK1, unlike STAT3 and AnxA2, is not a membrane-associated protein. Interestingly, the behavior of Prdx2 was very similar to that of the Prdx1 peroxidatic cysteine mutant (C52A) in that it could also only interact with ASK1 in cells. This suggests that there is a third protein mediating this interaction, which would bring Prdx1 C52A close enough to ASK1 for mLumin recombination without the actual formation of a Prdx1-ASK1 mixed disulfide, which does not occur according to our BLI results. The C52A BiFC result thus represents the amount of putative facilitation being accomplished by the unknown facilitator. To summarize more accurately and concisely: the Prdx1:ASK1 interaction is likely enhanced when a facilitator is present but is still able to occur without one. Moreover, even when the C52 of Prdx1 is mutated, if the facilitator is present (i.e., in cells), then it will bring Prdx1 in close proximity to ASK1 to encourage disulfide interaction even if chemically it is unable to occur because of the lack of the peroxidatic cysteine.
To explore which proteins can act as potential scaffolding proteins for the Prdx:ASK1 interaction, we searched the BioGrid database (version 4.3.196) to find common interactors between Prdx1, Prdx2, and ASK1 (
Figure 6). We found five such interactors, namely, cyclin-dependent kinase 2 (CDK2), egl-9 family hypoxia-inducible factor 3 (EGLN3), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), KIAA1429, and thioredoxin (Trx1).
From the five proteins in the overlap, Trx1 is the most likely candidate for the role of a mediator of the Prdx1 and Prdx2 interaction with ASK1, as it fits well with the known role of reduced Trx1 in binding ASK1 and inhibiting its kinase activity at basal H
2O
2 levels [
32]. Taken together, our results suggest the following model of the Prdx1:ASK1 interaction (
Figure 7), which is based on kinetic competition (i) for the binding to the ASK1-TBD: between C
P-C
R Prdx1 (or C
P-OH of Prdx1) and Trx1 (oxidized and reduced) [
32,
63] and (ii) for the binding to C
P-C
R Prdx1: between reduced Trx1 and ASK1-TBD. At basal H
2O
2 levels, the prevalent species of all the competitors is reduced Trx1, and ASK1-TBD is preferentially bound to it. Upon an increase in H
2O
2, Prdx1 and Prdx2 become oxidized (C
P-C
R) and are subsequently reduced by Trx1 bound to ASK1. As oxidized Trx1 binds ASK1-TBD with a lower affinity [
32] and structural changes in the ASK1-TBD make Trx1 binding unfavorable [
64], the chance for the exposed Cys250 of ASK1-TBD to perform a nucleophilic attack on oxidized Prdx1 increases (competition (i)). At the same time, oxidized Trx1 would not compete with Cys250 for C
P-C
R Prdx1 (competition (ii)). In case the ASK1-Prdx1 interaction proceeds via the intermolecular Prdx1 disulfide, this can either come from another Prdx1 dimer or from the other Prdx1 subunit of the Prdx1 dimer that oxidized the ASK1-bound Trx1. According to this model, the Trx1 bound to ASK1 acts as a recruitment platform for Prdx1 (or Prdx2) in the crowded environment of the cell. In vitro, where there is no need to recruit specific proteins, Trx1 is not needed.
The read-out of our BIFC experiments does not allow discrimination between Prdx1 that is oxidizing Trx1 bound to ASK1 or Cys250 of ASK1, as complementation has a distance range up to 10 nm [
49]. This would explain why we observe the Prdx2:ASK1 interaction in cells, but not in vitro: fluorescence complementation occurs when Prdx2 is oxidizing Trx1 bound to ASK1 yet does not in fact interact with ASK1 itself. In the case of the Prdx1 peroxidatic cysteine mutant, we can envisage a scenario where it would still bind to Trx1, as was observed for a peroxidatic cysteine mutant of a plant Prdx [
65], leading to fluorescence complementation. This interaction occurs exclusively upon treatment with H
2O
2, as mutant Prdx1 could still form a dimer with an endogenous Prdx1 (C
P-OH of the endogenous Prdx1 subunit would condense with the C
R of the peroxidatic cysteine mutant) that would oxidize the Trx1. An interaction with Trx1, rather than ASK1 itself, could potentially explain why the Prdx2 and Prdx1 C52A interaction with ASK1 yields lower fluorescence complementation than the Prdx1 WT and the Prdx1 C173A mutant: as the BiFC signal is cumulative, if complementation occurs both when Prdx1 interacts with Trx1 and ASK1, Prdx1 WT would yield higher signals. A repetition of the experiments in Prdx1 KO cells would help to clarify this. Regardless of the precise mechanism, the results of our in vitro data performed without Trx1 suggest that the role of Trx1 is limited to shielding the Cys250 of ASK1 at basal H
2O
2 levels and is not necessary for the Prdx1:ASK1 interaction. This Trx-shield is based on an estimated Trx1 concentration in HEK293 cells of 40–50 µM [
66] and, taking the K
D of reduced (0.3 ± 0.1 μM, [
32]) and oxidized (4 ± 2 μM, [
67]) Trx1 into account, oxidized Trx1 will directly dissociate from the TBD of ASK1. To see whether Trx1 is needed for ASK1 activation by Prdx1, experiments investigating the kinase activity of ASK1 would be necessary. These, however, are beyond the scope of the present study.
Alternatively, Prdx2 or the Prdx1 peroxidatic cysteine mutant could be interacting with domains other than the TBD, either covalently or non-covalently. To fully understand the Prdx2:ASK1 interaction, a separate study is required featuring various Prdx2 mutants and ASK1 domains using techniques such as co-IP and BiFC.
Another fate of the Prdx1 Cys
P-OH that was not discussed above is its hyperoxidation to Cys
P-O
2H. This sensitivity to hyperoxidation is notably variable between different isoforms. For example, Prdx1 is less prone to hyperoxidation than Prdx2 [
44]. The repair of sulfinylated Prdx1 can occur by Srx, which releases the repaired Prdx1 in the SOH form [
43]. This opens the possibility of Srx also acting as a scaffolding protein. We envisage that Srx could facilitate the Prdx1-ASK1 interaction both directly and indirectly. Respectively, it could be simply factoring into the amount of SOH available to participate in redox-relays or it could be serving as a scaffold. Moreover, the variability in hyperoxidation sensitivity could be a reason for scaffolding protein requirements for Prdx2 but not Prdx1.
Apart from the insights into the Prdx1:ASK1 interaction, this study also showed that constructs for BiFC can be selected based on an in silico approach using publicly available iTASSER and HADDOCK webservers, thus circumventing the laborious task of testing various combinations of BiFC pairs.
The discovery of AnxA2 as a scaffolding protein for Prdx2:STAT3 [
35] was hailed as one that brings us closer to solving the mystery of what dictates the specificity of Prdx redox-relays. The results of the investigation of the Prdx2:CRMP2 interaction then dampened those hopes, as they suggested that the organization of the same redox-relay was cell type-specific [
68]. The findings presented here, showing that scaffolding proteins are not needed for mediating all redox-relays, and that Prdx2 can interact with Prdx1 interactors, further suggest that the molecular details of redox-relays are more complex than we anticipated and that we are still a long way away from understanding how Prdxs and their targets find each other in the “molecularly crowded” environment of the cell. The overall role of peroxiredoxins and redox signaling in pathophysiology, however, guarantees that all efforts to disentangle the complexity of redox-relay organization will be fully justified.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}