
Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis

 ,
, 
_Haendeler.png)
Abstract
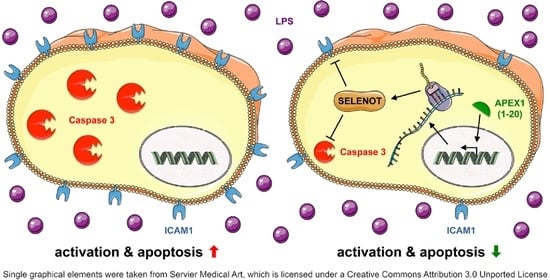
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cultivation of Primary Human Endothelial Cells and HEK293
2.2. Lentiviral Production and Transduction of EC
2.3. Isolation of Total Cellular RNA
2.4. RNA Sequencing and Bioinformatic Analysis
2.5. cDNA Synthesis
2.6. Polymerase Chain Reaction (PCR)
2.7. Plasmids
2.8. Transient Transfection of EC
2.9. Immunostaining of EC
2.10. Immunoblotting
2.11. Statistics
3. Results
3.1. APEX1(1-20) Induces Specific Transcriptome Changes in EC in Response to LPS
3.2. Expression of PXDN and SELENOT Is Specifically Upregulated after LPS Treatment of EC Expressing APEX1(1-20)
3.3. Generation of a SELENOT Expression Vector and Intracellular Localization of the Overexpressed Protein
3.4. SELENOT Overexpression Inhibits LPS-Induced Endothelial Cell Activation
3.5. SELENOT Overexpression Inhibits LPS-Induced Endothelial Cell Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Zhao, B.; Bowden, R.A.; Stavchansky, S.A.; Bowman, P.D. Human endothelial cell response to gram-negative lipopolysaccharide assessed with cDNA microarrays. Am. J. Physiol. Cell Physiol. 2001, 281, C1587–C1595. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Chan, H.; Wong, S.H.; Wang, M.H.; Yu, J.; Xiao, Z.; Liu, X.; Choi, G.; Leung, C.C.; Wong, W.T.; et al. The involvement of regulatory non-coding RNAs in sepsis: A systematic review. Crit. Care 2016, 20, 383. [Google Scholar] [CrossRef] [Green Version]
- Dyballa-Rukes, N.; Jakobs, P.; Eckers, A.; Ale-Agha, N.; Serbulea, V.; Aufenvenne, K.; Zschauer, T.C.; Rabanter, L.L.; Jakob, S.; von Ameln, F.; et al. The Anti-Apoptotic Properties of APEX1 in the Endothelium Require the First 20 Amino Acids and Converge on Thioredoxin-1. Antioxid. Redox Signal. 2017, 26, 616–629. [Google Scholar] [CrossRef]
- Ale-Agha, N.; Goy, C.; Jakobs, P.; Spyridopoulos, I.; Gonnissen, S.; Dyballa-Rukes, N.; Aufenvenne, K.; von Ameln, F.; Zurek, M.; Spannbrucker, T.; et al. CDKN1B/p27 is localized in mitochondria and improves respiration-dependent processes in the cardiovascular system-New mode of action for caffeine. PLoS Biol. 2018, 16, e2004408. [Google Scholar] [CrossRef]
- Goy, C.; Czypiorski, P.; Altschmied, J.; Jakob, S.; Rabanter, L.L.; Brewer, A.C.; Ale-Agha, N.; Dyballa-Rukes, N.; Shah, A.M.; Haendeler, J. The imbalanced redox status in senescent endothelial cells is due to dysregulated Thioredoxin-1 and NADPH oxidase 4. Exp. Gerontol. 2014, 56, 45–52. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Jakob, S.; Schroeder, P.; Lukosz, M.; Büchner, N.; Spyridopoulos, I.; Altschmied, J.; Haendeler, J. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J. Biol. Chem. 2008, 283, 33155–33161. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, P.; Popp, R.; Wiegand, B.; Altschmied, J.; Haendeler, J. Nuclear redox-signaling is essential for apoptosis inhibition in endothelial cells--important role for nuclear thioredoxin-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Cheng, G.; Salerno, J.C.; Cao, Z.; Pagano, P.J.; Lambeth, J.D. Identification and characterization of VPO1, a new animal heme-containing peroxidase. Free Radic. Biol. Med. 2008, 45, 1682–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Li, H.; Cao, Z.; Qiu, X.; McCormick, S.; Thannickal, V.J.; Nauseef, W.M. Vascular peroxidase-1 is rapidly secreted, circulates in plasma, and supports dityrosine cross-linking reactions. Free Radic. Biol. Med. 2011, 51, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Bhave, G.; Cummings, C.F.; Vanacore, R.M.; Kumagai-Cresse, C.; Ero-Tolliver, I.A.; Rafi, M.; Kang, J.S.; Pedchenko, V.; Fessler, L.I.; Fessler, J.H.; et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 2012, 8, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Medfai, H.; Khalil, A.; Rousseau, A.; Nuyens, V.; Paumann-Page, M.; Sevcnikar, B.; Furtmüller, P.G.; Obinger, C.; Moguilevsky, N.; Peulen, O.; et al. Human peroxidasin 1 promotes angiogenesis through ERK1/2, Akt, and FAK pathways. Cardiovasc. Res. 2019, 115, 463–475. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, H.K.; Naidansuren, P.; Ham, K.A.; Choi, H.S.; Ahn, H.Y.; Kim, M.; Kang, D.H.; Kang, S.W.; Joe, Y.A. Peroxidasin is essential for endothelial cell survival and growth signaling by sulfilimine crosslink-dependent matrix assembly. FASEB J. 2020, 34, 10228–10241. [Google Scholar] [CrossRef]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef]
- Pothion, H.; Jehan, C.; Tostivint, H.; Cartier, D.; Bucharles, C.; Falluel-Morel, A.; Boukhzar, L.; Anouar, Y.; Lihrmann, I. Selenoprotein T: An Essential Oxidoreductase Serving as a Guardian of Endoplasmic Reticulum Homeostasis. Antioxid. Redox Signal. 2020, 33, 1257–1275. [Google Scholar] [CrossRef]
- Boukhzar, L.; Hamieh, A.; Cartier, D.; Tanguy, Y.; Alsharif, I.; Castex, M.; Arabo, A.; El Hajji, S.; Bonnet, J.J.; Errami, M.; et al. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef]
- Huang, X.; Sun, B.; Zhang, J.; Gao, Y.; Li, G.; Chang, Y. Selenium Deficiency Induced Injury in Chicken Muscular Stomach by Downregulating Selenoproteins. Biol. Trace Elem. Res. 2017, 179, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bao, D.; Lei, C.T.; Tang, H.; Zhang, C.Y.; Su, H.; Zhang, C. Selenoprotein T protects against cisplatin-induced acute kidney injury through suppression of oxidative stress and apoptosis. FASEB J. 2020, 34, 11983–11996. [Google Scholar] [CrossRef]
- Addinsall, A.B.; Wright, C.R.; Andrikopoulos, S.; van der Poel, C.; Stupka, N. Emerging roles of endoplasmic reticulum-resident selenoproteins in the regulation of cellular stress responses and the implications for metabolic disease. Biochem. J. 2018, 475, 1037–1057. [Google Scholar] [CrossRef]
- Tujebajeva, R.M.; Copeland, P.R.; Xu, X.M.; Carlson, B.A.; Harney, J.W.; Driscoll, D.M.; Hatfield, D.L.; Berry, M.J. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000, 1, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J.; Messmer, U.K.; Brüne, B.; Neugebauer, E.; Dimmeler, S. Endotoxic shock leads to apoptosis in vivo and reduces Bcl-2. Shock 1996, 6, 405–409. [Google Scholar] [CrossRef]
- Pober, J.S. Endothelial activation: Intracellular signaling pathways. Arthritis Res. Ther. 2002, 4 (Suppl. 3), S109–S116. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Haendeler, J.; Rippmann, V.; Nehls, M.; Zeiher, A.M. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996, 399, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Carlson, B.A.; Xu, X.M.; Gladyshev, V.N.; Hatfield, D.L. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005, 280, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Droz, B.; Greve, P.; Gottschalk, P.; Poffet, D.; McGrath, S.P.; Seneviratne, S.I.; Smith, P.; Winkel, L.H. Selenium deficiency risk predicted to increase under future climate change. Proc. Natl. Acad. Sci. USA 2017, 114, 2848–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes Junior, E.; Leite, H.P.; Konstantyner, T. Selenium and selenoproteins: From endothelial cytoprotection to clinical outcomes. Transl. Res. 2019, 208, 85–104. [Google Scholar] [CrossRef]
- Mertens, K.; Lowes, D.A.; Webster, N.R.; Talib, J.; Hall, L.; Davies, M.J.; Beattie, J.H.; Galley, H.F. Low zinc and selenium concentrations in sepsis are associated with oxidative damage and inflammation. Br. J. Anaesth. 2015, 114, 990–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jing, J.; Yan, H.; Tang, J.; Jia, G.; Liu, G.; Chen, X.; Tian, G.; Cai, J.; Shang, H.; et al. Selenium Pretreatment Alleviated LPS-Induced Immunological Stress Via Upregulation of Several Selenoprotein Encoding Genes in Murine RAW264.7 Cells. Biol. Trace Elem. Res. 2018, 186, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, B.; Cao, H.L.; Wang, R.Y.; Lu, Z.Y.; Chi, R.F.; Li, B. Selenium Supplementation Protects Against Lipopolysaccharide-Induced Heart Injury via Sting Pathway in Mice. Biol. Trace Elem. Res. 2021, 199, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Boukhzar, L.; Granieri, M.C.; Alsharif, I.; Mazza, R.; Lefranc, B.; Tota, B.; Leprince, J.; Cerra, M.C.; Anouar, Y.; et al. A selenoprotein T-derived peptide protects the heart against ischaemia/reperfusion injury through inhibition of apoptosis and oxidative stress. Acta Physiol. 2018, 223, e13067. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, I.; Boukhzar, L.; Lefranc, B.; Godefroy, D.; Aury-Landas, J.; Rego, J.D.; Rego, J.D.; Naudet, F.; Arabo, A.; Chagraoui, A.; et al. Cell-penetrating, antioxidant SELENOT mimetic protects dopaminergic neurons and ameliorates motor dysfunction in Parkinson’s disease animal models. Redox Biol. 2021, 40, 101839. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merk, D.; Ptok, J.; Jakobs, P.; von Ameln, F.; Greulich, J.; Kluge, P.; Semperowitsch, K.; Eckermann, O.; Schaal, H.; Ale-Agha, N.; et al. Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants 2021, 10, 1427. https://doi.org/10.3390/antiox10091427
Merk D, Ptok J, Jakobs P, von Ameln F, Greulich J, Kluge P, Semperowitsch K, Eckermann O, Schaal H, Ale-Agha N, et al. Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants. 2021; 10(9):1427. https://doi.org/10.3390/antiox10091427
Chicago/Turabian StyleMerk, Dennis, Johannes Ptok, Philipp Jakobs, Florian von Ameln, Jan Greulich, Pia Kluge, Kathrin Semperowitsch, Olaf Eckermann, Heiner Schaal, Niloofar Ale-Agha, and et al. 2021. "Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis" Antioxidants 10, no. 9: 1427. https://doi.org/10.3390/antiox10091427
APA StyleMerk, D., Ptok, J., Jakobs, P., von Ameln, F., Greulich, J., Kluge, P., Semperowitsch, K., Eckermann, O., Schaal, H., Ale-Agha, N., Altschmied, J., & Haendeler, J. (2021). Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants, 10(9), 1427. https://doi.org/10.3390/antiox10091427