
Metabolic Adaptions/Reprogramming in Islet Beta-Cells in Response to Physiological Stimulators—What Are the Consequences




Abstract
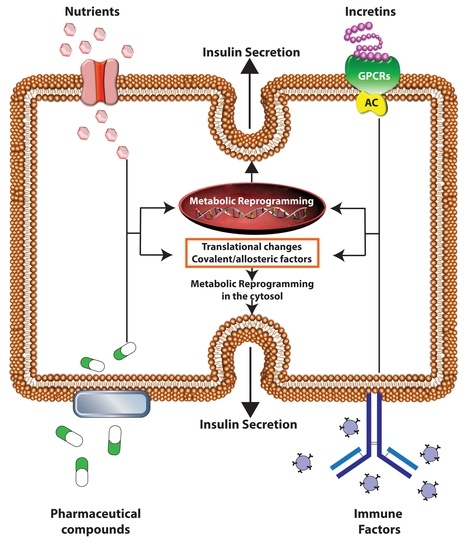
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Evidence for Nutrient Regulation of Metabolic Adaptions/Reprogramming in β-Cells
2.1. Glucose
2.2. Lipids
3. Evidence for Immune Driven Metabolic Adaptions in β-Cells, with a Focus on Increased Oxidative and Endoplasmic Reticulum Stress
4. Evidence for Endocrine Regulation of Metabolic Adaptions/Reprogramming in β-Cells
5. Evidence for Pharmaceutical Drug Induced Metabolic Adaptions/Reprogramming in β-Cells
5.1. Antidiabetic Medications and Pancreatic β-Cell Metabolism
5.2. Pancreatic β-Cell G-Protein Coupled Receptors and Cell Metabolism
5.3. Contribution of Insulin, and Therapeutic Availability, to Metabolic Function of β-Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newsholme, P.; Cruzat, V.; Arfuso, F.; Keane, K. Nutrient regulation of insulin secretion and action. J. Endocrinol. 2014, 221, R105–R120. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Fujimoto, S.; Tsuura, Y.; Mukai, E.; Takeda, T.; Hamamoto, Y.; Takehiro, M.; Fujita, J.; Yamada, Y.; Seino, Y. Ouabain suppresses glucose-induced mitochondrial ATP production and insulin release by generating reactive oxygen species in pancreatic islets. Diabetes 2002, 51, 2522–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorsman, P.; Renström, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Rustenbeck, I.; Schulze, T.; Morsi, M.; Alshafei, M.; Panten, U. What Is the Metabolic Amplification of Insulin Secretion and Is It (Still) Relevant? Metabolites 2021, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Gembal, M.; Gilon, P.; Henquin, J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Investg. 1992, 89, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Kelly, B.; O’neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ma, R.; Wu, Y.; Zhai, Y.; Li, S. Reciprocal regulation of metabolic reprogramming and epigenetic modifications in cancer. Front. Genet. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife 2019, 8, e50163. [Google Scholar] [CrossRef] [PubMed]
- Cliff, T.S.; Dalton, S. Metabolic switching and cell fate decisions: Implications for pluripotency, reprogramming and development. Curr. Opin. Genet. Dev. 2017, 46, 44–49. [Google Scholar]
- Sato, Y.; Aizawa, T.; Komatsu, M.; Okada, N.; Yamada, T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes 1992, 41, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Nagashima, K.; Tashiro, F.; Kotake, K.; Yoshitomi, H.; Tamamoto, A.; Gonoi, T.; Iwanaga, T.; Miyazaki, J.; Seino, S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 10402–10406. [Google Scholar] [CrossRef] [Green Version]
- Remedi, M.S.; Rocheleau, J.V.; Tong, A.; Patton, B.L.; McDaniel, M.L.; Piston, D.W.; Koster, J.C.; Nichols, C.G. Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia 2006, 49, 2368–2378. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Holness, M.J. The pyruvate carboxylase-pyruvate dehydrogenase axis in islet pyruvate metabolism: Going round in circles? Islets 2011, 3, 302–319. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Simoes, D.; Riva, P.; Peliciari-Garcia, R.A.; Cruzat, V.F.; Graciano, M.F.; Munhoz, A.C.; Taneda, M.; Cipolla-Neto, J.; Carpinelli, A.R. Melatonin modifies basal and stimulated insulin secretion via NADPH oxidase. J. Endocrinol. 2016, 231, 235–244. [Google Scholar] [CrossRef]
- McClenaghan, N.H.; Scullion, S.M.; Mion, B.; Hewage, C.; Malthouse, J.P.; Flatt, P.R.; Newsholme, P.; Brennan, L. Prolonged L-alanine exposure induces changes in metabolism, Ca(2+) handling and desensitization of insulin secretion in clonal pancreatic beta-cells. Clin. Sci. 2009, 116, 341–351. [Google Scholar] [CrossRef]
- Schulze, T.; Morsi, M.; Reckers, K.; Bruning, D.; Seemann, N.; Panten, U.; Rustenbeck, I. Metabolic amplification of insulin secretion is differentially desensitized by depolarization in the absence of exogenous fuels. Metabolism 2017, 67, 1–13. [Google Scholar] [CrossRef]
- Sanchez, P.K.M.; Khazaei, M.; Gatineau, E.; Geravandi, S.; Lupse, B.; Liu, H.; Dringen, R.; Wojtusciszyn, A.; Gilon, P.; Maedler, K. LDHA is enriched in human islet alpha cells and upregulated in type 2 diabetes. Biochem. Biophys. Res. Commun. 2021, 568, 158–166. [Google Scholar] [CrossRef]
- Maassen, J.A.; t Hart, L.M.; Janssen, G.M.; Reiling, E.; Romijn, J.A.; Lemkes, H.H. Mitochondrial diabetes and its lessons for common Type 2 diabetes. Biochem. Soc. Trans. 2006, 34, 819–823. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Bender, K.; Kiely, A.; Brennan, L. Amino acid metabolism, insulin secretion and diabetes. Biochem. Soc. Trans. 2007, 35, 1180–1186. [Google Scholar] [CrossRef]
- Marmol, P.; Pardo, B.; Wiederkehr, A.; Del Arco, A.; Wollheim, C.B.; Satrustegui, J. Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J. Biol. Chem. 2009, 284, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, K.; Maechler, P.; McClenaghan, N.H.; Flatt, P.R.; Newsholme, P. Overexpression of the malate-aspartate NADH shuttle member Aralar1 in the clonal beta-cell line BRIN-BD11 enhances amino-acid-stimulated insulin secretion and cell metabolism. Clin. Sci. 2009, 117, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spégel, P.; Mulder, H. Metabolomics analysis of nutrient metabolism in β-cells. J. Mol. Biol. 2020, 432, 1429–1445. [Google Scholar] [CrossRef] [PubMed]
- Spégel, P.; Sharoyko, V.V.; Goehring, I.; Danielsson, A.P.; Malmgren, S.; Nagorny, C.L.; Andersson, L.E.; Koeck, T.; Sharp, G.W.; Straub, S.G. Time-resolved metabolomics analysis of β-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem. J. 2013, 450, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, E.P.; Procopio, J.; Carvalho, C.R.; Carpinelli, A.R.; Newsholme, P.; Curi, R. New insights into fatty acid modulation of pancreatic beta-cell function. Int. Rev. Cytol. 2006, 248, 1–41. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic beta cells and insulin sensitive cells and tissues—Importance to cell metabolism, function and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 137, C420–C433. [Google Scholar] [CrossRef]
- Jezek, P.; Jaburek, M.; Holendova, B.; Plecita-Hlavata, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Carlessi, R.; Walz, N.; Cruzat, V.F.; Keane, K.; John, A.N.; Jiang, F.X.; Carnagarin, R.; Dass, C.R.; Newsholme, P. Pigment epithelium-derived factor (PEDF) regulates metabolism and insulin secretion from a clonal rat pancreatic beta cell line BRIN-BD11 and mouse islets. Mol. Cell. Endocrinol. 2016, 426, 50–60. [Google Scholar] [CrossRef]
- Cantley, J.; Davenport, A.; Vetterli, L.; Nemes, N.J.; Whitworth, P.T.; Boslem, E.; Thai, L.M.; Mellett, N.; Meikle, P.J.; Hoehn, K.L.; et al. Disruption of beta cell acetyl-CoA carboxylase-1 in mice impairs insulin secretion and beta cell mass. Diabetologia 2019, 62, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curi, R.; Newsholme, P.; Marzuca-Nassr, G.N.; Takahashi, H.K.; Hirabara, S.M.; Cruzat, V.; Krause, M.; de Bittencourt, P.I., Jr. Regulatory principles in metabolism-then and now. Biochem. J. 2016, 473, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Corkey, B.E.; Madiraju, S.R.M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 2020, 63, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Seiron, P.; Wiberg, A.; Kuric, E.; Krogvold, L.; Jahnsen, F.L.; Dahl-Jorgensen, K.; Skog, O.; Korsgren, O. Characterisation of the endocrine pancreas in type 1 diabetes: Islet size is maintained but islet number is markedly reduced. J. Pathol. Clin. Res. 2019, 5, 248–255. [Google Scholar] [CrossRef]
- Calella, P.; Galle, F.; Fornelli, G.; Liguori, G.; Valerio, G. Type 1 diabetes and body composition in youth: A systematic review. Diabetes Metab. Res. Rev. 2020, 36, e3211. [Google Scholar] [CrossRef]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and beta-Cell Dysfunction. Oxid. Med. Cell Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [Green Version]
- Cruzat, V.F.; Keane, K.N.; Scheinpflug, A.L.; Cordeiro, R.; Soares, M.J.; Newsholme, P. Alanyl-glutamine improves pancreatic beta-cell function following ex vivo inflammatory challenge. J. Endocrinol. 2015, 224, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.; Clemente-Casares, X.; Revelo, X.S.; Winer, S.; Winer, D.A. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes 2015, 64, 1886–1897. [Google Scholar] [CrossRef] [Green Version]
- Khawandanah, J. Double or hybrid diabetes: A systematic review on disease prevalence, characteristics and risk factors. Nutr. Diabetes 2019, 9, 33. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.-M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, J.W.; Steiner, S.R.; O’Neill, C.M.; Nunemaker, C.S. The central role of calcium in the effects of cytokines on beta-cell function: Implications for type 1 and type 2 diabetes. Cell Calcium 2011, 50, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to β-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R.; Smith, P.; Ashcroft, F. Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic β-cells. Biochem. J. 1993, 294, 35–42. [Google Scholar] [CrossRef]
- Kindmark, H.; Köhler, M.; Brown, G.; Branstrom, R.; Larsson, O.; Berggren, P.-O. Glucose-induced oscillations in cytoplasmic free Ca2+ concentration precede oscillations in mitochondrial membrane potential in the pancreatic β-cell. J. Biol. Chem. 2001, 276, 34530–34536. [Google Scholar] [CrossRef] [Green Version]
- Krippeit-Drews, P.; Düfer, M.; Drews, G. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem. Biophys. Res. Commun. 2000, 267, 179–183. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabe, D.; Seino, Y. Two incretin hormones GLP-1 and GIP: Comparison of their actions in insulin secretion and β cell preservation. Prog. Biophys. Mol. Biol. 2011, 107, 248–256. [Google Scholar] [CrossRef]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.J.; Rosenkilde, M.M. GIP as a therapeutic target in diabetes and obesity: Insight from incretin co-agonists. J. Clin. Endocrinol. Metab. 2020, 105, e2710–e2716. [Google Scholar]
- Bailey, C.J. GIP analogues and the treatment of obesity-diabetes. Peptides 2020, 125, 170202. [Google Scholar] [CrossRef]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.D.; Finan, B.; Bloom, S.; D’Alessio, D.; Drucker, D.J.; Flatt, P.; Fritsche, A.; Gribble, F.; Grill, H.; Habener, J. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Kang, G.X.; Leech, C.A.; Chepurny, O.G.; Coetzee, W.A.; Holz, G.G. Role of the cAMP sensor Epac as a determinant of K-ATP channel ATP sensitivity in human pancreatic beta-cells and rat INS-1 cells. J. Physiol. 2008, 586, 1307–1319. [Google Scholar] [CrossRef]
- Kang, G.; Joseph, J.W.; Chepurny, O.G.; Monaco, M.; Wheeler, M.B.; Bos, J.L.; Schwede, F.; Genieser, H.G.; Holz, G.G. Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J. Biol. Chem. 2003, 278, 8279–8285. [Google Scholar] [CrossRef] [Green Version]
- Doyle, M.E.; Egan, J.M. Mechanisms of Action of GLP-1 in the Pancreas. Pharmacol. Ther. 2007, 113, 546–593. [Google Scholar] [CrossRef] [Green Version]
- El, K.; Gray, S.; Capozzi, M.; Knuth, E.; Jin, E.; Svendsen, B.; Clifford, A.; Brown, J.; Encisco, S.; Chazotte, B. GIP mediates the incretin effect and glucose tolerance by dual actions on α cells and β cells. Sci. Adv. 2021, 7, eabf1948. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Dattaroy, D.; Pham, J.; Wang, L.; Barella, L.F.; Cui, Y.; Wilkins, K.J.; Roth, B.L.; Hochgeschwender, U.; Matschinsky, F.M. Intraislet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 2019, 4, e127994. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, M.E.; Wait, J.B.; Jepchumba Koech, A.N.G.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when β cells are active. JCI Insight 2019, 4, e129954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.P.; An, Z.; Wagner, C.; Lewis, A.G.; Cohen, E.B.; Li, B.; Mahbod, P.; Sandoval, D.; Perez-Tilve, D.; Tamarina, N. The role of β cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. 2014, 19, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.P.; Sorrell, J.E.; Haller, A.; Roelofs, K.; Hutch, C.R.; Kim, K.-S.; Gutierrez-Aguilar, R.; Li, B.; Drucker, D.J.; D’Alessio, D.A. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab. 2017, 25, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Whalley, N.; Pritchard, L.; Smith, D.; White, A. Processing of proglucagon to GLP-1 in pancreatic a-cells: Is this a paracrine mechanism enabling GLP-1 to act on b-cells. J. Endocrinol. 2011, 211, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buteau, J.; Foisy, S.; Joly, E.; Prentki, M. Glucagon-Like Peptide 1 Induces Pancreatic β-Cell Proliferation Via Transactivation of the Epidermal Growth Factor Receptor. Diabetes 2003, 52, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.E.; Ussher, J.R.; Mulvihill, E.E.; Kolic, J.; Baggio, L.L.; Cao, X.; Liu, Y.; Lamont, B.J.; Morii, T.; Streutker, C.J. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016, 22, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.-H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP-and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Dong, X.; Fisher, T.L.; Dunn, S.; Omer, A.K.; Weir, G.; White, M.F. Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J. Biol. Chem. 2006, 281, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, S.; Spinas, G.; Maedler, K.; Zuellig, R.; Lehmann, R.; Donath, M.; Trüb, T.; Niessen, M. Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human β-cells from hyperglycemia-induced apoptosis. Exp. Cell Res. 2005, 303, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Lingohr, M.K.; Dickson, L.M.; Wrede, C.E.; Briaud, I.; McCuaig, J.F.; Myers Jr, M.G.; Rhodes, C.J. Decreasing IRS-2 expression in pancreatic β-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol. Cell. Endocrinol. 2003, 209, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Sampaio, A.V.; Underhill, T.M.; McIntosh, C.H. Noncanonical activation of Akt/protein kinase B in β-cells by the incretin hormone glucose-dependent insulinotropic polypeptide. J. Biol. Chem. 2009, 284, 10764–10773. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Nian, C.; Widenmaier, S.; McIntosh, C.H. Glucose-dependent insulinotropic polypeptide-mediated up-regulation of β-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol. Cell. Biol. 2008, 28, 1644. [Google Scholar] [CrossRef] [Green Version]
- Hui, H.; Nourparvar, A.; Zhao, X.; Perfetti, R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A-and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology 2003, 144, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Li, X.; Gu, X.; Chan, J.; Xu, G. Exendin-4 protects pancreatic beta cells from human islet amyloid polypeptide-induced cell damage: Potential involvement of AKT and mitochondria biogenesis. Diabetes Obes. Metab. 2010, 12, 815–824. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Ao, Z.; Kim, S.-J.; Warnock, G.; McIntosh, C.H. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the β-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J. Biol. Chem. 2009, 284, 30372–30382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-H.; Kim, E.-H.; Jung, H.S.; Yang, D.; Park, E.-Y.; Jun, H.-S. EX4 stabilizes and activates Nrf2 via PKCδ, contributing to the prevention of oxidative stress-induced pancreatic beta cell damage. Toxicol. Appl. Pharmacol. 2017, 315, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Millán, E.; Martín, M.; Goya, L.; Lizárraga-Mollinedo, E.; Escrivá, F.; Ramos, S.; Álvarez, C. Glucagon-like peptide-1 improves beta-cell antioxidant capacity via extracellular regulated kinases pathway and Nrf2 translocation. Free Radic. Biol. Med. 2016, 95, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Storace, D.; Durante, A.; Odetti, P.; Viviani, G. Glucagon-like peptide-1 counteracts the detrimental effects of Advanced Glycation End-Products in the pancreatic beta cell line HIT-T 15. Biochem. Biophys. Res. Commun. 2010, 398, 462–466. [Google Scholar] [CrossRef]
- Shimoda, M.; Kanda, Y.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Matsuki, M.; Kaku, K. The human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells via regulation of cell kinetics and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetologia 2011, 54, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Fang, Q.-H.; Cui, Y.-T.; Shen, Q.-L.; Liu, Q.; Wang, P.-H.; Yu, D.-M.; Li, C.-J. Liraglutide prevents β-cell apoptosis via inactivation of NOX2 and its related signaling pathway. J. Diabetes Complicat. 2019, 33, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.-J.; Krause, K.-H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef] [Green Version]
- Friedrichsen, B.N.; Neubauer, N.; Lee, Y.C.; Gram, V.K.; Blume, N.; Petersen, J.S.; Nielsen, J.H.; Møldrup, A. Stimulation of pancreatic β-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J. Endocrinol. 2006, 188, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Pelech, S.L.; Pederson, R.A.; McIntosh, C.H. Glucose-dependent insulinotropic polypeptide activates the Raf-Mek1/2-ERK1/2 module via a cyclic AMP/cAMP-dependent protein kinase/Rap1-mediated pathway. J. Biol. Chem. 2002, 277, 37088–37097. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, X.; Li, L.-X.; Brubaker, P.L.; Edlund, H.; Drucker, D.J. β-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 2005, 54, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Cornu, M.; Modi, H.; Kawamori, D.; Kulkarni, R.N.; Joffraud, M.; Thorens, B. Glucagon-like peptide-1 increases beta-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J. Biol. Chem. 2010, 285, 10538–10545. [Google Scholar] [CrossRef] [Green Version]
- Cornu, M.; Yang, J.Y.; Jaccard, E.; Poussin, C.; Widmann, C.; Thorens, B. Glucagon-like peptide-1 protects beta-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes 2009, 58, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar]
- Rowlands, J.; Cruzat, V.; Carlessi, R.; Newsholme, P. Insulin and IGF-1 receptor autocrine loops are not required for Exendin-4 induced changes to pancreatic β-cell bioenergetic parameters and metabolism in BRIN-BD11 cells. Peptides 2018, 100, 140–149. [Google Scholar] [CrossRef]
- Carlessi, R.; Chen, Y.; Rowlands, J.; Cruzat, V.F.; Keane, K.N.; Egan, L.; Mamotte, C.; Stokes, R.; Gunton, J.E.; Bittencourt, P.I.H.; et al. GLP-1 receptor signalling promotes beta-cell glucose metabolism via mTOR-dependent HIF-1alpha activation. Sci. Rep. 2017, 7, 2661. [Google Scholar] [CrossRef]
- Stamateris, R.E.; Sharma, R.B.; Kong, Y.; Ebrahimpour, P.; Panday, D.; Ranganath, P.; Zou, B.; Levitt, H.; Parambil, N.A.; O’Donnell, C.P. Glucose induces mouse β-cell proliferation via IRS2, MTOR, and cyclin D2 but not the insulin receptor. Diabetes 2016, 65, 981–995. [Google Scholar] [CrossRef] [Green Version]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Winter, K.; Nian, C.; Tsuneoka, M.; Koda, Y.; McIntosh, C.H. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic β-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 2005, 280, 22297–22307. [Google Scholar]
- Trumper, A.; Trumper, K.; Horsch, D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta (INS-1)-cells. J. Endocrinol. 2002, 174, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional loss of pancreatic islets in type 2 diabetes: How can we halt it? Metabolism 2020, 110, 154304. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, Z.; Zhang, C.; Cai, Z.; Zhang, J. Metformin, beyond an insulin sensitizer, targeting heart and pancreatic β cells. Biochim. Biophys. Acta Mol. Bas. Dis. 2017, 1863, 1984–1990. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Messaoudi, S.; Rongen, G.A.; de Boer, R.A.; Riksen, N.P. The cardioprotective effects of metformin. Curr. Opin. Lipidol. 2011, 22, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277. [Google Scholar] [CrossRef]
- Leclerc, I.; Woltersdorf, W.W.; da Silva Xavier, G.; Rowe, R.L.; Cross, S.E.; Korbutt, G.S.; Rajotte, R.V.; Smith, R.; Rutter, G.A. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: Impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1023–E1031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investg. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Hashemitabar, M.; Bahramzadeh, S.; Saremy, S.; Nejaddehbashi, F. Glucose plus metformin compared with glucose alone on β-cell function in mouse pancreatic islets. Biomed. Rep. 2015, 3, 721–725. [Google Scholar] [CrossRef]
- Kitabchi, A.E.; Temprosa, M.; Knowler, W.C.; Kahn, S.E.; Fowler, S.E.; Haffner, S.M.; Andres, R.; Saudek, C.; Edelstein, S.L.; Arakaki, R.; et al. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: Effects of lifestyle intervention and metformin. Diabetes 2005, 54, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Lupi, R.; Del Guerra, S.; Fierabracci, V.; Marselli, L.; Novelli, M.; Patanè, G.; Boggi, U.; Mosca, F.; Piro, S.; Del Prato, S.; et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 2002, 51 (Suppl. S1), S134–S137. [Google Scholar] [CrossRef] [Green Version]
- Lupi, R.; Del Guerra, S.; Tellini, C.; Giannarelli, R.; Coppelli, A.; Lorenzetti, M.; Carmellini, M.; Mosca, F.; Navalesi, R.; Marchetti, P. The biguanide compound metformin prevents desensitization of human pancreatic islets induced by high glucose. Eur. J. Pharmacol. 1999, 364, 205–209. [Google Scholar] [CrossRef]
- Patanè, G.; Piro, S.; Rabuazzo, A.M.; Anello, M.; Vigneri, R.; Purrello, F. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: A direct metformin effect on pancreatic beta-cells. Diabetes 2000, 49, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Masini, M.; Anello, M.; Bugliani, M.; Marselli, L.; Filipponi, F.; Boggi, U.; Purrello, F.; Occhipinti, M.; Martino, L.; Marchetti, P.; et al. Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio. Diabetes Res. Clin. Pract. 2014, 104, 163–170. [Google Scholar] [CrossRef]
- González-Barroso, M.M.; Anedda, A.; Gallardo-Vara, E.; Redondo-Horcajo, M.; Rodríguez-Sánchez, L.; Rial, E. Fatty acids revert the inhibition of respiration caused by the antidiabetic drug metformin to facilitate their mitochondrial β-oxidation. Biochim. Biophys. Acta 2012, 1817, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Deat. Dis. 2011, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Lee, M.W.; Lee, Y.J.; Kim, S.M. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem. Biophys. Res. Commun. 2012, 417, 147–152. [Google Scholar] [CrossRef]
- Simon-Szabó, L.; Kokas, M.; Mandl, J.; Kéri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.L.; Huang, S.L.; Leng, Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms. Int. J. Biol. Sci. 2015, 11, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic Aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef]
- Modak, M.A.; Parab, P.B.; Ghaskadbi, S.S. Control of hyperglycemia significantly improves oxidative stress profile of pancreatic islets. Islets 2011, 3, 234–240. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, C.M.; Docherty, K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001, 44, 1203–1214. [Google Scholar] [CrossRef]
- Jara, M.A.; Werneck-De-Castro, J.P.; Lubaczeuski, C.; Johnson, J.D.; Bernal-Mizrachi, E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to β-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 2020, 12, 32–40. [Google Scholar] [CrossRef]
- Obafemi, T.O.; Jaiyesimi, K.F.; Olomola, A.A.; Olasehinde, O.R.; Olaoye, O.A.; Adewumi, F.D.; Afolabi, B.A.; Adewale, O.B.; Akintayo, C.O.; Ojo, O.A. Combined effect of metformin and gallic acid on inflammation, antioxidant status, endoplasmic reticulum (ER) stress and glucose metabolism in fructose-fed streptozotocin-induced diabetic rats. Toxicol. Rep. 2021, 8, 1419–1427. [Google Scholar] [CrossRef]
- Pan, Q.R.; Li, W.H.; Wang, H.; Sun, Q.; Xiao, X.H.; Brock, B.; Schmitz, O. Glucose, metformin, and AICAR regulate the expression of G protein-coupled receptor members in INS-1 beta cell. Horm. Metab. Res. 2009, 41, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kieffer, T.J. New aspects of an old drug: Metformin as a glucagon-like peptide 1 (GLP-1) enhancer and sensitiser. Diabetologia 2011, 54, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-α in mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef]
- Brown, J.B.; Conner, C.; Nichols, G.A. Secondary failure of metformin monotherapy in clinical practice. Diabetes Care 2010, 33, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef] [Green Version]
- Matthews, D.A.-O.; Del Prato, S.; Mohan, V.; Mathieu, C.; Vencio, S.; Chan, J.C.N.; Stumvoll, M.; Paldánius, P.M. Insights from VERIFY: Early Combination Therapy Provides Better Glycaemic Durability Than a Stepwise Approach in Newly Diagnosed Type 2 Diabetes. Diabetes Ther. 2020, 11, 2465–2476. [Google Scholar] [CrossRef]
- Ball, A.J.; Flatt, P.R.; McClenaghan, N.H. Desensitization of sulphonylurea- and nutrient-induced insulin secretion following prolonged treatment with glibenclamide. Eur. J. Pharmacol. 2000, 408, 327–333. [Google Scholar] [CrossRef]
- Maedler, K.; Carr, R.D.; Bosco, D.; Zuellig, R.A.; Berney, T.; Donath, M.Y. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Cui, X.; Feng, J.; Gu, L.; Lang, S.; Wei, T.; Yang, J.; Liu, J.; Le, Y.; Wang, H.; et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism 2020, 111, 154324. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Bouchard, R. Dipeptidyl peptidase-4 inhibition in diabetic rats leads to activation of the transcription factor CREB in β-cells. Eur. J. Pharmacol. 2015, 755, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Delobel, M.; Dalle, S. G-protein–coupled receptors controlling pancreatic β-cell functional mass for the treatment of type 2 diabetes. Curr. Opin. Endocr. Metab. Res. 2021, 16, 113–118. [Google Scholar] [CrossRef]
- Winzell, M.S.; Ahrén, B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol. Ther. 2007, 116, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Persaud, S.J. Islet G-protein coupled receptors: Therapeutic potential for diabetes. Curr. Opin. Pharmacol. 2017, 37, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Rossi, M.; Cohen, A.; Pham, J.; Zheng, H.; Dattaroy, D.; Mukaibo, T.; Melvin, J.E.; Langel, J.L.; Hattar, S.; et al. Allosteric modulation of β-cell M(3) muscarinic acetylcholine receptors greatly improves glucose homeostasis in lean and obese mice. Proc. Natl. Acad. Sci. USA 2019, 116, 18684–18690. [Google Scholar] [CrossRef] [Green Version]
- Usui, R.; Yabe, D.; Fauzi, M.; Goto, H.; Botagarova, A.; Tokumoto, S.; Tatsuoka, H.; Tahara, Y.; Kobayashi, S.; Manabe, T.; et al. GPR40 activation initiates store-operated Ca(2+) entry and potentiates insulin secretion via the IP3R1/STIM1/Orai1 pathway in pancreatic β-cells. Sci. Rep. 2019, 9, 15562. [Google Scholar] [CrossRef] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Tudurí, E.; Imbernon, M.; Hernández-Bautista, R.J.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202. [Google Scholar] [CrossRef]
- McKillop, A.M.; Moran, B.M.; Abdel-Wahab, Y.H.; Flatt, P.R. Evaluation of the insulin releasing and antihyperglycaemic activities of GPR55 lipid agonists using clonal beta-cells, isolated pancreatic islets and mice. Br. J. Pharmacol. 2013, 170, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Romero-Zerbo, S.Y.; Rafacho, A.; Diaz-Arteaga, A.; Suarez, J.; Quesada, I.; Imbernon, M.; Ross, R.A.; Dieguez, C.; Fonseca, F.R.d.; Nogueiras, R. A role for the putative cannabinoid receptor GPR55 in the islets of Langerhans. J. Endocrinol. 2011, 211, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Song, S.; Ruz-Maldonado, I.; Pingitore, A.; Huang, G.C.; Baker, D.; Jones, P.M.; Persaud, S.J. GPR55-dependent stimulation of insulin secretion from isolated mouse and human islets of Langerhans. Diabetes Obes. Metab. 2016, 18, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, B.M.; Flatt, P.R.; McKillop, A.M. G protein-coupled receptors: Signalling and regulation by lipid agonists for improved glucose homoeostasis. Acta Diabetol. 2016, 53, 177–188. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, A.G.; Miskelly, M.G.; Moore, C.B.T.; Nesbit, M.A.; Christie, K.A.; Owolabi, A.I.; Flatt, P.R.; McKillop, A.M. CRISPR/Cas9 gene editing demonstrates metabolic importance of GPR55 in the modulation of GIP release and pancreatic beta cell function. Peptides 2020, 125, 170251. [Google Scholar] [CrossRef]
- Simcocks, A.C.; O’Keefe, L.; Jenkin, K.A.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. A potential role for GPR55 in the regulation of energy homeostasis. Drug Discov. Today 2014, 19, 1145–1151. [Google Scholar] [CrossRef]
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.; Hoi, M.P.M. G-protein coupled receptor 55 agonists increase insulin secretion through inositol trisphosphate-mediated calcium release in pancreatic β-cells. Eur. J. Pharmacol. 2019, 854, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, I.B.; Leibiger, B.; Berggren, P.O. Insulin signaling in the pancreatic beta-cell. Annu. Rev. Nutr. 2008, 28, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Aspinwall, C.A.; Lakey, J.R.; Kennedy, R.T. Insulin-stimulated insulin secretion in single pancreatic beta cells. J. Biol. Chem. 1999, 274, 6360–6365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.D.; Misler, S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human beta cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14566–14571. [Google Scholar] [CrossRef] [Green Version]
- Luciani, D.S.; Johnson, J.D. Acute effects of insulin on beta-cells from transplantable human islets. Mol. Cell Endocrinol. 2005, 241, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Goforth, P.B.; Zhang, M.; Satin, L.S. Insulin activates ATP-sensitive K(+) channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 2001, 50, 2192–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagren, O.I.; Tengholm, A. Glucose and insulin synergistically activate phosphatidylinositol 3-kinase to trigger oscillations of phosphatidylinositol 3,4,5-trisphosphate in beta-cells. J. Biol. Chem. 2006, 281, 39121–39127. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.C.; Plant, T.D.; Gilon, P.; Detimary, P.; Nenquin, M.; Henquin, J.C. Multiple effects and stimulation of insulin secretion by the tyrosine kinase inhibitor genistein in normal mouse islets. Br. J. Pharmacol. 1995, 114, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawalich, W.S.; Zawalich, K.C. Effects of glucose, exogenous insulin, and carbachol on C-peptide and insulin secretion from isolated perifused rat islets. J. Biol. Chem. 2002, 277, 26233–26237. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newsholme, P.; Rowlands, J.; Rose’Meyer, R.; Cruzat, V. Metabolic Adaptions/Reprogramming in Islet Beta-Cells in Response to Physiological Stimulators—What Are the Consequences. Antioxidants 2022, 11, 108. https://doi.org/10.3390/antiox11010108
Newsholme P, Rowlands J, Rose’Meyer R, Cruzat V. Metabolic Adaptions/Reprogramming in Islet Beta-Cells in Response to Physiological Stimulators—What Are the Consequences. Antioxidants. 2022; 11(1):108. https://doi.org/10.3390/antiox11010108
Chicago/Turabian StyleNewsholme, Philip, Jordan Rowlands, Roselyn Rose’Meyer, and Vinicius Cruzat. 2022. "Metabolic Adaptions/Reprogramming in Islet Beta-Cells in Response to Physiological Stimulators—What Are the Consequences" Antioxidants 11, no. 1: 108. https://doi.org/10.3390/antiox11010108
APA StyleNewsholme, P., Rowlands, J., Rose’Meyer, R., & Cruzat, V. (2022). Metabolic Adaptions/Reprogramming in Islet Beta-Cells in Response to Physiological Stimulators—What Are the Consequences. Antioxidants, 11(1), 108. https://doi.org/10.3390/antiox11010108