
Role of FOXO3a Transcription Factor in the Regulation of Liver Oxidative Injury


Abstract
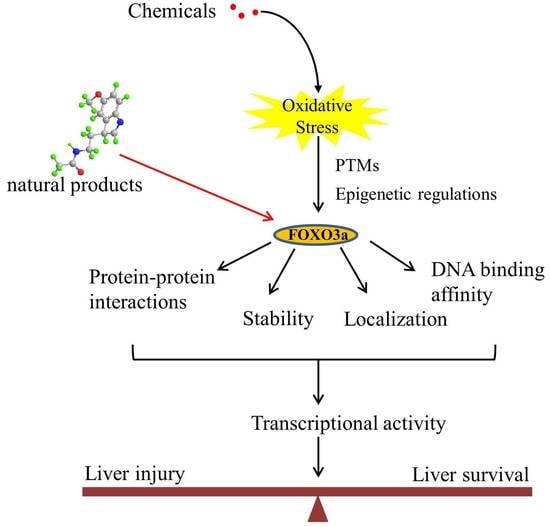
:
1. Introduction
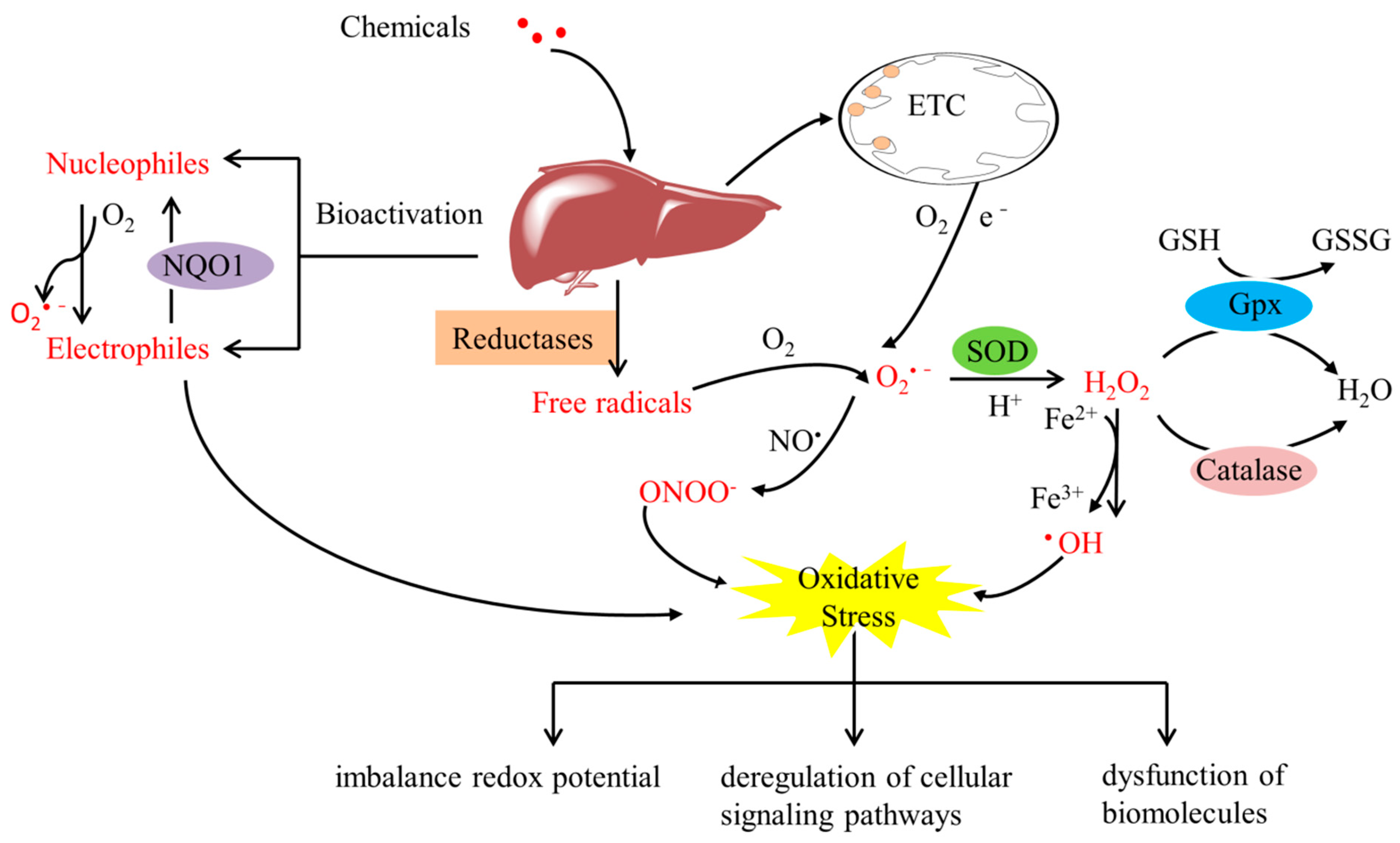
2. Architecture of FOXO3a Domains
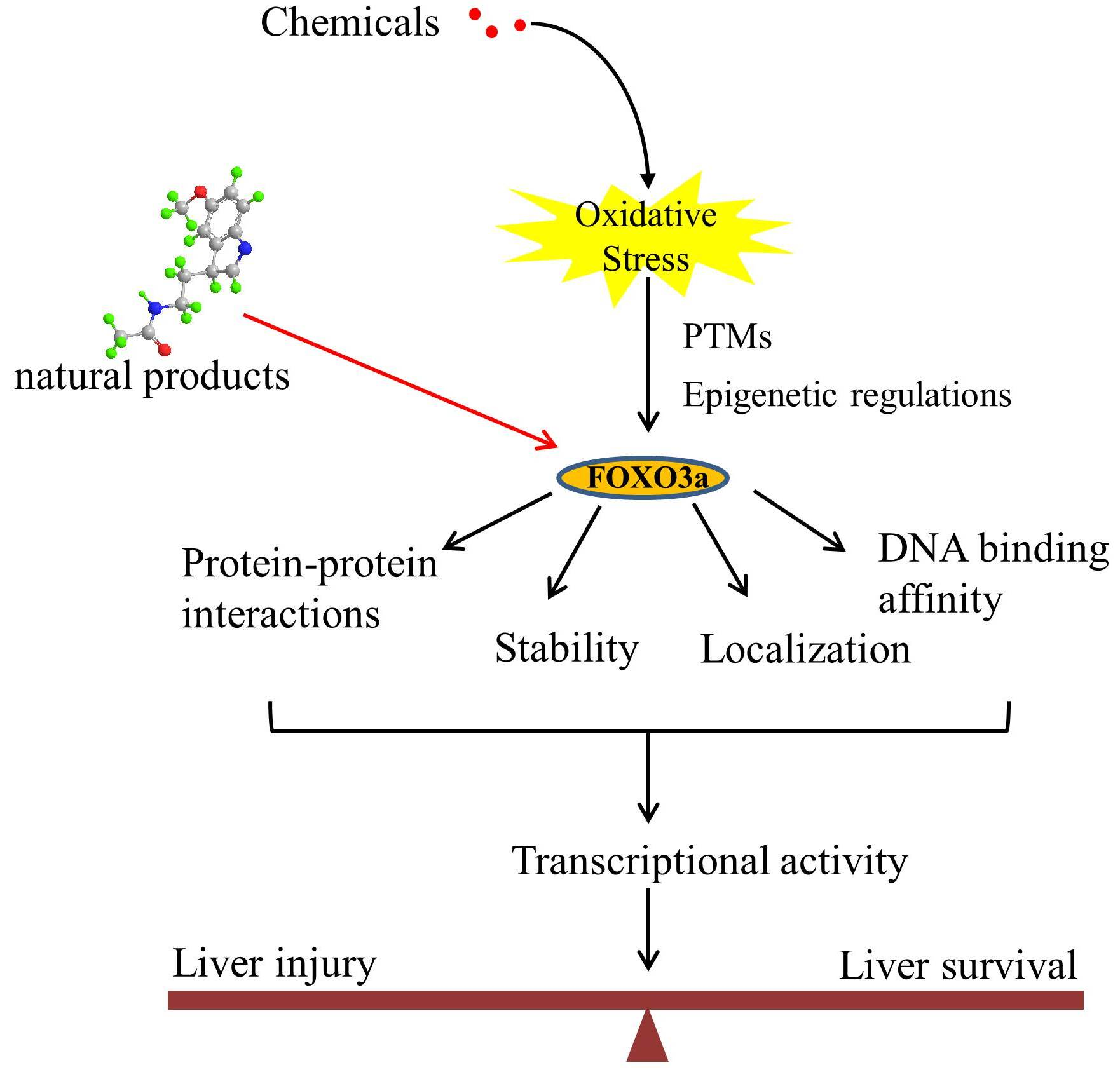
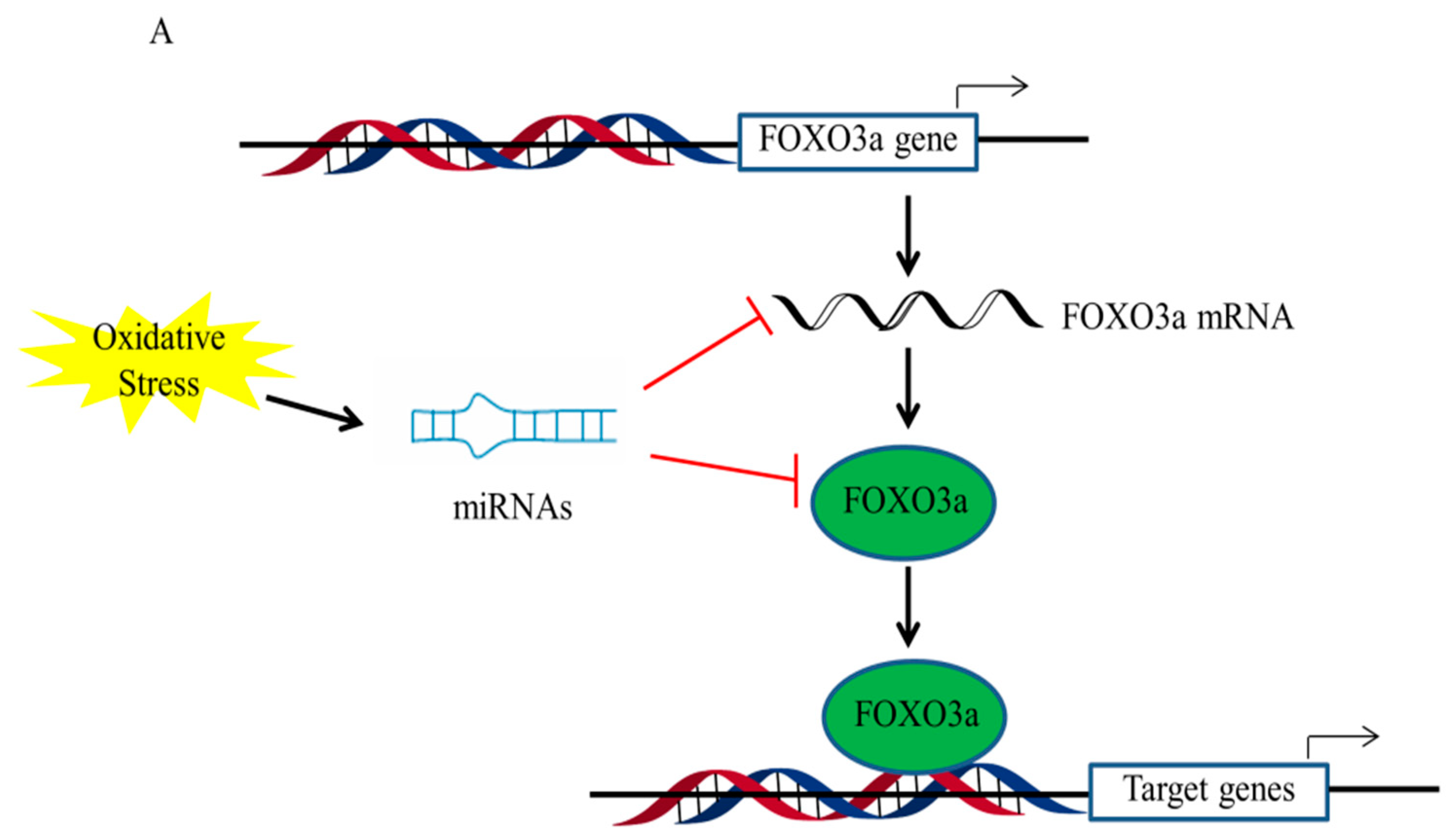
3. Regulation of FOXO3a upon Oxidative Stress
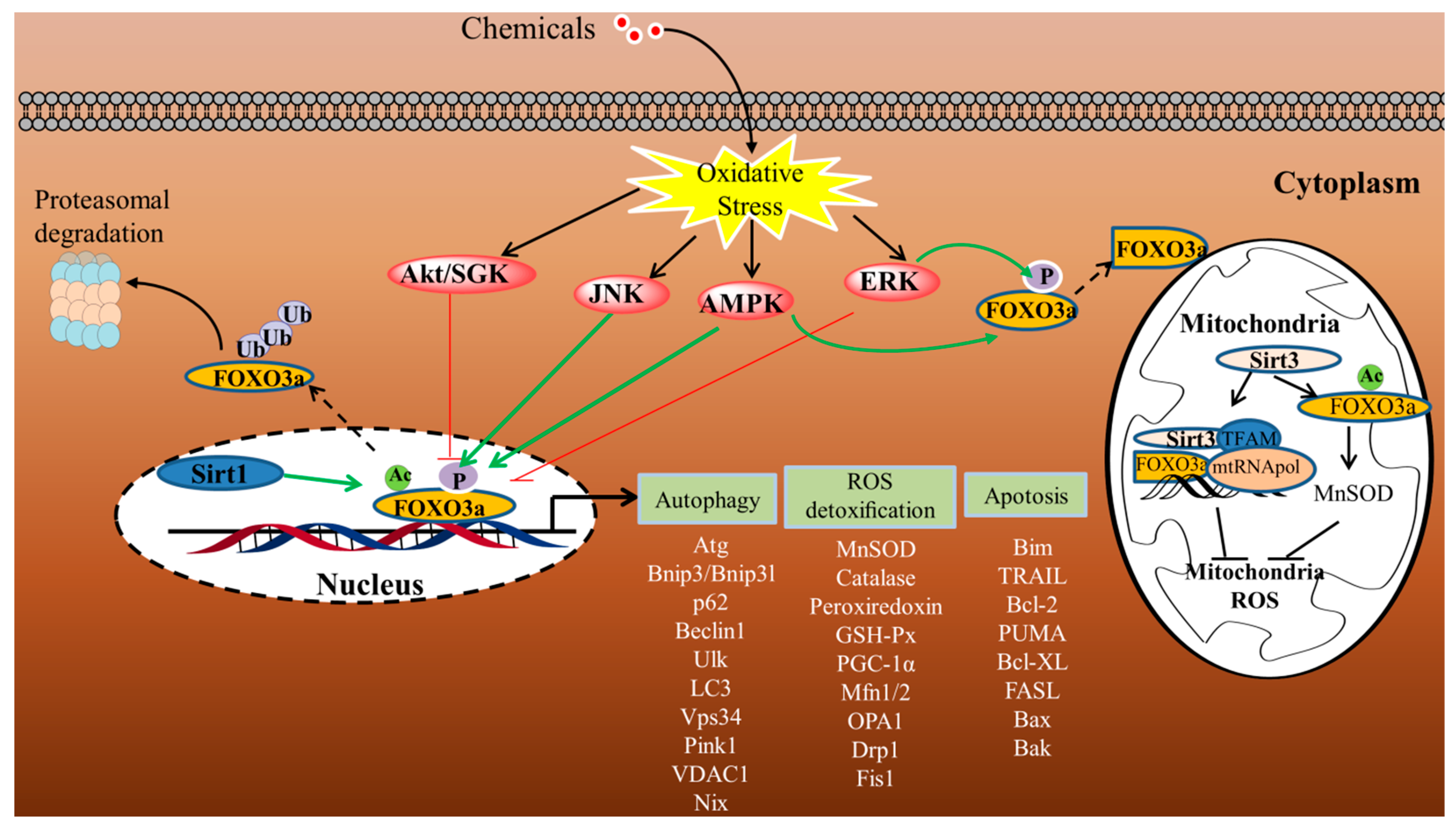
3.1. Post-Translational Modifications
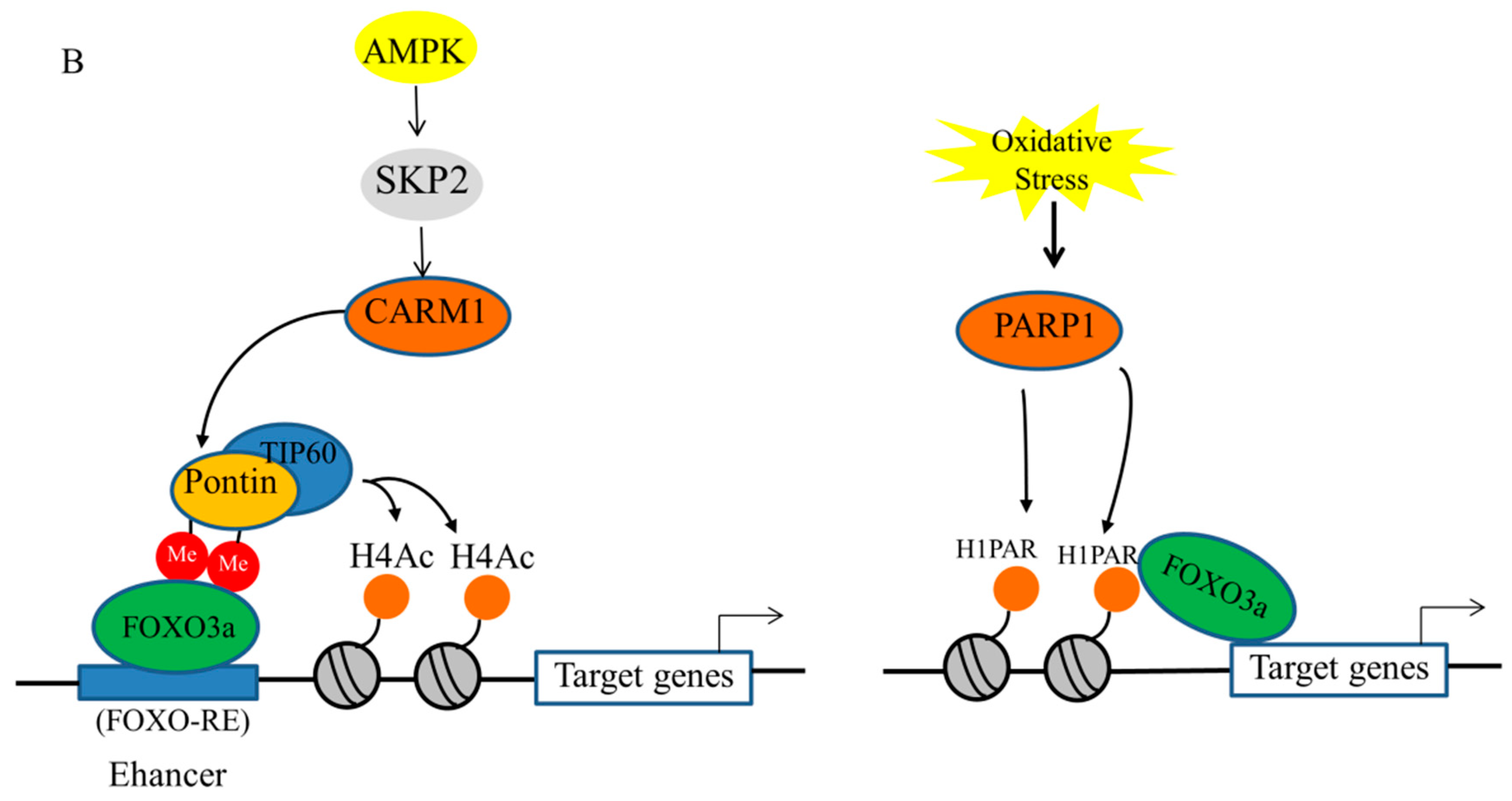
3.2. Epigenetic Regulation
4. Role of FOXO3a in the Liver
4.1. FOXO3a-Autophagy Axis
4.2. FOXO3a-Regulated Apoptosis
4.3. FOXO3a-Mediated Regulation of the Antioxidant System
5. FOXO3a as a Potential Therapeutic Target
6. Conclusions and Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Manautou, J.E. Molecular mechanisms underlying chemical liver injury. Expert. Rev. Mol. Med. 2012, 14, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplowitz, N. Biochemical and cellular mechanisms of toxic liver injury. Semin Liver Dis. 2002, 22, 137–144. [Google Scholar] [CrossRef]
- Donato, M.; Tolosa, L. High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells. Antioxidants 2021, 10, 106. [Google Scholar] [CrossRef]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar]
- Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells 2019, 8, 1110. [Google Scholar] [CrossRef]
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, e2972. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, J.; Zhang, S.; Weinman, S.A. FOXO3-dependent apoptosis limits alcohol-induced liver inflammation by promoting infiltrating macrophage differentiation. Cell Death Dis. 2018, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Puerto, M.C.; Verhagen, L.P.; Braat, A.K.; Lam, E.W.; Coffer, P.J.; Lorenowicz, M.J. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy 2016, 12, 1804–1816. [Google Scholar] [CrossRef]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Tikhanovich, I.; Cox, J.; Weinman, S.A. Forkhead box class O transcription factors in liver function and disease. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S1), 125–131. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.K.; Stringham, B.A.; Mohr, A.M.; Wehrkamp, C.J.; Lu, S.; Phillippi, M.A.; Harrison-Findik, D.; Mott, J.L. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J. Lipid Res. 2017, 58, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Cao, Q.; Wang, F.; Huang, L.Y.; Sang, T.T.; Liu, F.; Chen, S.Y. SIRT1 Protects Against Oxidative Stress-Induced Endothelial Progenitor Cells Apoptosis by Inhibiting FOXO3a via FOXO3a Ubiquitination and Degradation. J. Cell Physiol. 2015, 230, 2098–2107. [Google Scholar] [CrossRef]
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000, 349 Pt 2, 629–634. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Marshall, C.B.; Ikura, M. Forkhead followed by disordered tail: The intrinsically disordered regions of FOXO3a. Intrinsically Disord. Proteins 2015, 3, e1056906. [Google Scholar] [CrossRef] [PubMed]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Lepore Signorile, M.; Longo, G.; De Rasmo, D. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nho, R.S.; Hergert, P. FoxO3a and disease progression. World J. Biol. Chem. 2014, 5, 346–354. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Link, W. Introduction to FOXO Biology. Methods Mol Biol. 2019, 1890, 1–9. [Google Scholar]
- Williams, J.A.; Manley, S.; Ding, W.X. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J. Gastroenterol. 2014, 20, 12908–12933. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Mauriz, J.L.; García-Palomo, A.; González-Gallego, J. FoxO proteins: Regulation and molecular targets in liver cancer. Curr. Med. Chem. 2014, 21, 1231–1246. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, T.; Huang, P. Post-translational modifications of FOXO family proteins (Review). Mol. Med. Rep. 2016, 14, 4931–4941. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and acetylation modifications of FOXO3a: Independently or synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Niu, Y.; Wan, A.; Chen, D.; Liang, H.; Chen, X.; Sun, L.; Zhan, S.; Chen, L.; Cheng, C. RNA m6 A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated Autophagy. EMBO J. 2020, 39, e103181. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Venditti, P. Evolution of the Knowledge of Free Radicals and Other Oxidants. Oxid. Med. Cell. Longev. 2020, 2020, 9829176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wang, X.; Ma, L.; Deng, A.; Wang, S.; Chen, X. FoxO3 transcription factor promotes autophagy after transient cerebral ischemia/reperfusion. Int. J. Neurosci. 2019, 129, 738–745. [Google Scholar] [CrossRef]
- Luo, J. Autophagy and ethanol neurotoxicity. Autophagy 2014, 10, 2099–2108. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 2011, 14, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Thévenod, F.; Lee, W.K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 1743–1786. [Google Scholar] [CrossRef]
- Fujiki, K.; Inamura, H.; Matsuoka, M. Phosphorylation of FOXO3a by PI3K/Akt pathway in HK-2 renal proximal tubular epithelial cells exposed to cadmium. Arch. Toxicol. 2013, 87, 2119–2127. [Google Scholar] [CrossRef]
- Guo, H.; Yang, H.; Chen, H.; Li, W.; Tang, J.; Cheng, P.; Xie, Y.; Liu, Y.; Ding, G.; Cui, D.; et al. Molecular mechanisms of human thyrocyte dysfunction induced by low concentrations of polychlorinated biphenyl 118 through the Akt/FoxO3a/NIS pathway. J. Appl. Toxicol. 2015, 35, 992–998. [Google Scholar] [CrossRef]
- Yang, H.; Chen, H.; Guo, H.; Li, W.; Tang, J.; Xu, B.; Sun, M.; Ding, G.; Jiang, L.; Cui, D.; et al. Molecular mechanisms of 2, 3’, 4, 4’, 5-pentachlorobiphenyl-induced thyroid dysfunction in FRTL-5 cells. PLoS ONE 2015, 10, e0120133. [Google Scholar] [CrossRef]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120 Pt 15, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Li, C.; Li, L.; Liu, X.; Zhou, L.; Zhang, W.; Ni, S.; Lu, Y.; Chen, L.; Jeong, L.S.; et al. Neddylation Inactivation Facilitates FOXO3a Nuclear Export to Suppress Estrogen Receptor Transcription and Improve Fulvestrant Sensitivity. Clin. Cancer Res. 2019, 25, 3658–3672. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar]
- Vucicevic, L.; Misirkic, M.; Ciric, D.; Martinovic, T.; Jovanovic, M.; Isakovic, A.; Markovic, I.; Saponjic, J.; Foretz, M.; Rabanal-Ruiz, Y.; et al. Transcriptional block of AMPK-induced autophagy promotes glutamate excitotoxicity in nutrient-deprived SH-SY5Y neuroblastoma cells. Cell. Mol. Life Sci. 2020, 77, 3383–3399. [Google Scholar] [CrossRef]
- Rosso, P.; Fioramonti, M.; Fracassi, A. AMPK in the central nervous system: Physiological roles and pathological implications. Res. Rep. Biol. 2016, 7, 1–13. [Google Scholar]
- Li, X.N.; Song, J.; Zhang, L.; LeMaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef] [Green Version]
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krähenbühl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215. [Google Scholar] [CrossRef]
- Marsin, A.S.; Bouzin, C.; Bertrand, L.; Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 2002, 277, 30778–30783. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- de Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential changes in the signal transduction responses of skeletal muscle following food deprivation. FASEB J. 2006, 20, 2579–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol- induced liver disease. Biochem. Pharmacol. 2013, 86, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Candau, R.; Bernardi, H. AMP-activated protein kinase stabilizes FOXO3 in primary myotubes. Biochem. Biophys. Res. Commun. 2018, 499, 493–498. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar]
- Wang, X.; Liu, J.Z.; Hu, J.X.; Wu, H.; Li, Y.L.; Chen, H.L.; Bai, H.; Hai, C.X. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic. Biol. Med. 2011, 51, 539–551. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, J.; Yang, M.J.; Hong, S.P.; Lee, C.K.; Kubota, Y.; Lim, D.S.; Koh, G.Y. A MST1-FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat. Commun. 2019, 10, 838. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Tian, J.; Zhou, D.; Chen, L. Mst1 and Mst2 kinases: Regulations and diseases. Cell Biosci. 2013, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, L.; Wu, Y.; Su, D.; Wang, N.; Wang, J.; Shi, C.; Lv, L.; Zhang, S. Tanshinol suppresses inflammatory factors in a rat model of vascular dementia and protects LPS-treated neurons via the MST1-FOXO3 signaling pathway. Brain Res. 2016, 1646, 304–314. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, S.; Cui, M.; Gao, X.; Sun, D.; Qin, X.; Narsinh, K.; Li, C.; Jia, H.; Li, C.; et al. Rosuvastatin enhances the therapeutic efficacy of adipose-derived mesenchymal stem cells for myocardial infarction via PI3K/Akt and MEK/ERK pathways. Basic Res. Cardiol. 2013, 108, 333. [Google Scholar] [CrossRef]
- Bei, Y.; Pan, L.L.; Zhou, Q.; Zhao, C.; Xie, Y.; Wu, C.; Meng, X.; Gu, H.; Xu, J.; Zhou, L.; et al. Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med. 2019, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Dolloff, N.G.; El-Deiry, W.S. ERK and MDM2 prey on FOXO3a. Nat. Cell Biol. 2008, 10, 125–126. [Google Scholar] [CrossRef]
- Wang, X.; Chen, W.R.; Xing, D. A pathway from JNK through decreased ERK and Akt activities for FOXO3a nuclear translocation in response to UV irradiation. J. Cell Physiol. 2012, 227, 1168–1178. [Google Scholar] [CrossRef]
- Li, Z.; Bridges, B.; Olson, J.; Weinman, S.A. The interaction between acetylation and serine-574 phosphorylation regulates the apoptotic function of FOXO3. Oncogene 2017, 36, 1887–1898. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, J.; Tikhanovich, I.; Kuravi, S.; Helzberg, J.; Dorko, K.; Roberts, B.; Kumer, S.; Weinman, S.A. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ. 2016, 23, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ye, M.; Bucur, O.; Zhu, S.; Tanya Santos, M.; Rabinovitz, I.; Wei, W.; Gao, D.; Hahn, W.C.; Khosravi-Far, R. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol. Biol. Cell. 2010, 21, 1140–1152. [Google Scholar] [CrossRef] [Green Version]
- Nho, R.S.; Kahm, J. beta1-Integrin-collagen interaction suppresses FoxO3a by the coordination of Akt and PP2A. J. Biol. Chem. 2010, 285, 14195–14209. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Zhao, J.; Peng, W.; Wu, H.; Zhang, Y. Resveratrol rescues cadmium-induced mitochondrial injury by enhancing transcriptional regulation of PGC-1α and SOD2 via the Sirt3/FoxO3a pathway in TCMK-1 cells. Biochem. Biophys. Res. Commun. 2017, 486, 198–204. [Google Scholar] [CrossRef]
- He, X.; Gao, J.; Hou, H.; Qi, Z.; Chen, H.; Zhang, X.X. Inhibition of Mitochondrial Fatty Acid Oxidation Contributes to Development of Nonalcoholic Fatty Liver Disease Induced by Environmental Cadmium Exposure. Environ. Sci. Technol. 2019, 53, 13992–14000. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Kashiwagi, E.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Seki, N.; Naito, S. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer Sci. 2010, 101, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Nguyen, T.T.; Santamaria, A.; Bowman, A.B.; Buha Djordjevic, A.; Paoliello, M.M.B.; Skalny, A.V.; Aschner, M. Sirtuins as molecular targets, mediators, and protective agents in metal-induced toxicity. Arch. Toxicol. 2021, 95, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Y.; Zhu, G.; Liang, Y.; Liu, B.; Wu, Y.; Han, M.; Sun, W.; Han, Y.; Chen, G.; et al. SIRT1 downregulation mediated Manganese-induced neuronal apoptosis through activation of FOXO3a-Bim/PUMA axis. Sci. Total Environ. 2019, 646, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, J.; Li, A.P.; Zhang, Z.L.; Li, Z.R.; Guo, K.J.; Zhao, K.C.; Ruan, Q.; Guo, L. Bone marrow mesenchymal stem cells could reduce the toxic effects of hexavalent chromium on the liver by decreasing endoplasmic reticulum stress-mediated apoptosis via SIRT1/HIF-1α signaling pathway in rats. Toxicol. Lett. 2019, 310, 31–38. [Google Scholar] [CrossRef]
- Fu, W.; Ma, Q.; Chen, L.; Li, P.; Zhang, M.; Ramamoorthy, S.; Nawaz, Z.; Shimojima, T.; Wang, H.; Yang, Y.; et al. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 2009, 284, 13987–14000. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Jeong, H.J.; Kim, H.; Choi, D.; Cho, S.C.; Seong, J.K.; Koo, S.H.; Kang, J.S. Skeletal muscle-specific Prmt1 deletion causes muscle atrophy via deregulation of the PRMT6-FOXO3 axis. Autophagy 2019, 15, 1069–1081. [Google Scholar] [CrossRef]
- Gupta, S.; Kadumuri, R.V.; Singh, A.K.; Chavali, S.; Dhayalan, A. Structure, Activity and Function of the Protein Arginine Methyltransferase 6. Life 2021, 11, 951. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; Zhang, Y.; Zhang, F.; Huang, K. PARP1 promote autophagy in cardiomyocytes via modulating FoxO3a transcription. Cell Death Dis. 2018, 9, 1047. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Kim, H.; Oh, S.; Lee, J.G.; Kee, M.; Ko, H.J.; Kweon, M.N.; Won, K.J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of Autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Zhao, S.; Yan, F.; Cheng, J.; Huang, L.; Chen, H.; Liu, Q.; Ji, X.; Yuan, Z. HDAC2 selectively regulates FOXO3a-mediated gene transcription during oxidative stress-induced neuronal cell death. J. Neurosci. 2015, 35, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Feng, L.; Zheng, Y.L.; Lu, J.; Fan, S.H.; Shan, Q.; Zheng, G.H.; Wang, Y.J.; Wu, D.M.; Li, M.Q.; et al. 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47) induces mitochondrial dysfunction and related liver injury via eliciting miR-34a-5p-mediated mitophagy impairment. Environ. Pollut. 2020, 258, 113693. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Voisin, S.; Russell, A.; Lamon, S. Recent advances in understanding the role of FOXO3. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, E.; Tang, Y.; Mao, J.; Shen, J.; Zheng, X.; Xie, S.; Zhang, S.; Wu, Y.; Liu, H.; et al. miR-223 overexpression inhibits doxorubicin-induced autophagy by targeting FOXO3a and reverses chemoresistance in hepatocellular carcinoma cells. Cell Death Dis. 2019, 10, 843. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, E10849–E10858. [Google Scholar] [CrossRef]
- Langlet, F.; Tarbier, M.; Haeusler, R.A.; Camastra, S.; Ferrannini, E.; Friedländer, M.R.; Accili, D. microRNA-205-5p is a modulator of insulin sensitivity that inhibits FOXO function. Mol. Metab. 2018, 17, 49–60. [Google Scholar] [CrossRef]
- Yu, Y.S.; Shin, H.R.; Kim, D.; Baek, S.A.; Choi, S.A.; Ahn, H.; Shamim, A.; Kim, J.; Kim, I.S.; Kim, K.K.; et al. Pontin arginine methylation by CARM1 is crucial for epigenetic regulation of Autophagy. Nat. Commun. 2020, 11, 6297. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell. 2015, 58, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Tao, Z.; Zheng, L.D.; Smith, C.; Luo, J.; Robinson, A.; Almeida, F.A.; Wang, Z.; Olumi, A.F.; Liu, D.; Cheng, Z. Estradiol signaling mediates gender difference in visceral adiposity via Autophagy. Cell Death Dis. 2018, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z. The FoxO-Autophagy Axis in Health and Disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zou, P.; Zheng, L.; Linarelli, L.E.; Amarell, S.; Passaro, A.; Liu, D.; Cheng, Z. Tamoxifen reduces fat mass by boosting reactive oxygen species. Cell Death Dis. 2015, 6, e1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, S.; Shen, Z.; Song, F. Autophagy and acetaminophen-induced hepatotoxicity. Arch. Toxicol. 2018, 92, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, T.; Rahman, S.U.; Hao, Q.; Li, W.; Liu, Z.; Ali Shah, F.; Murtaza, I.; Zhang, Z.; Yang, X.; Liu, G.; et al. Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FOXO3a regulation. J. Pineal Res. 2020, 69, e12667. [Google Scholar] [CrossRef]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [Green Version]
- Manley, S.; Ni, H.M.; Williams, J.A.; Kong, B.; DiTacchio, L.; Guo, G.; Ding, W.X. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol. 2014, 2, 991–1002. [Google Scholar] [CrossRef] [Green Version]
- Nepal, S.; Park, P.H. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: Involvement of AMPK/FoxO3A axis. Biochim. Biophys. Acta 2013, 1833, 2111–2125. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Xiong, X.; Tao, R.; DePinho, R.A.; Dong, X.C. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem. 2012, 287, 39107–39114. [Google Scholar] [CrossRef] [Green Version]
- Chaves, I.; van der Horst, G.T.; Schellevis, R.; Nijman, R.M.; Koerkamp, M.G.; Holstege, F.C.; Smidt, M.P.; Hoekman, M.F. Insulin-FOXO3 signaling modulates circadian rhythms via regulation of clock transcription. Curr. Biol. 2014, 24, 1248–1255. [Google Scholar] [CrossRef] [Green Version]
- Ferrell, J.M.; Chiang, J.Y. Circadian rhythms in liver metabolism and disease. Acta Pharm. Sin. B 2015, 5, 113–122. [Google Scholar] [CrossRef]
- Liu, B.; Yu, H.; Baiyun, R.; Lu, J.; Li, S.; Bing, Q.; Zhang, X.; Zhang, Z. Protective effects of dietary luteolin against mercuric chloride-induced lung injury in mice: Involvement of AKT/Nrf2 and NF-κB pathways. Food Chem. Toxicol. 2018, 113, 296–302. [Google Scholar] [CrossRef]
- Yang, F.; Pei, R.; Zhang, Z.; Liao, J.; Yu, W.; Qiao, N.; Han, Q.; Li, Y.; Hu, L.; Guo, J.; et al. Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicol. Vitr. 2019, 54, 310–316. [Google Scholar] [CrossRef]
- Tao, G.Z.; Lehwald, N.; Jang, K.Y.; Baek, J.; Xu, B.; Omary, M.B.; Sylvester, K.G. Wnt/β-catenin signaling protects mouse liver against oxidative stress-induced apoptosis through the inhibition of forkhead transcription factor FoxO3. J. Biol. Chem. 2013, 288, 17214–17224. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Han, B.; Li, S.; Wang, X.; Wu, P.; Liu, Y.; Li, J.; Han, B.; Deng, N.; Zhang, Z. The link between deacetylation and hepatotoxicity induced by exposure to hexavalent chromium. J. Adv. Res. 2021, 35, 129–140. [Google Scholar] [CrossRef]
- Huang, C.K.; Yu, T.; de la Monte, S.M.; Wands, J.R.; Derdak, Z.; Kim, M. Restoration of Wnt/β-catenin signaling attenuates alcoholic liver disease progression in a rat model. J. Hepatol. 2015, 63, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Barreyro, F.J.; Kobayashi, S.; Bronk, S.F.; Werneburg, N.W.; Malhi, H.; Gores, G.J. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 2007, 282, 27141–27154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Anati, L.; Kadekar, S.; Högberg, J.; Stenius, U. PCB153, TCDD and estradiol compromise the benzo[a]pyrene-induced p53-response via FoxO3a. Chem. Biol. Interact. 2014, 219, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, B.H. Drug Induced Liver Injury (DILI). Mechanisms and Medicinal Chemistry Avoidance/Mitigation Strategies. J. Med. Chem. 2020, 63, 11397–11419. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Ai, C.X.; Zhang, J.S.; Li, W.C. Pre-hypoxia exposure inhibited copper toxicity by improving energy metabolism, antioxidant defence and mitophagy in the liver of the large yellow croaker Larimichthys crocea. Sci. Total Environ. 2020, 708, 134961. [Google Scholar] [CrossRef]
- Yan, M.; Jin, S.; Liu, Y.; Wang, L.; Wang, Z.; Xia, T.; Chang, Q. Cajaninstilbene Acid Ameliorates Acetaminophen-Induced Liver Injury Through Enhancing Sestrin2/AMPK-Mediated Mitochondrial Quality Control. Front. Pharmacol. 2022, 13, 824138. [Google Scholar] [CrossRef]
- Barbier-Torres, L.; Iruzubieta, P.; Fernández-Ramos, D.; Delgado, T.C.; Taibo, D.; Guitiérrez-de-Juan, V.; Varela-Rey, M.; Azkargorta, M.; Navasa, N.; Fernández-Tussy, P.; et al. The mitochondrial negative regulator MCJ is a therapeutic target for acetaminophen-induced liver injury. Nat. Commun. 2017, 8, 2068. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Kim, A.D.; Kang, K.A.; Piao, M.J.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Hyun, C.L.; Kang, H.K.; Lee, N.H.; et al. Cytoprotective effect of eckol against oxidative stress-induced mitochondrial dysfunction: Involvement of the FoxO3a/AMPK pathway. J. Cell. Biochem. 2014, 115, 1403–1411. [Google Scholar] [CrossRef]
- Wang, J.; He, Y.; Peng, X.; Bo, L.v.; Wang, Z.; Song, Q. Characterization of cadmium-responsive transcription factors in wolf spider Pardosa pseudoannulata. Chemosphere 2021, 268, 129239. [Google Scholar] [CrossRef]
- Song, C.; Zhao, J.; Fu, B.; Li, D.; Mao, T.; Peng, W.; Wu, H.; Zhang, Y. Melatonin-mediated upregulation of Sirt3 attenuates sodium fluoride-induced hepatotoxicity by activating the MT1-PI3K/AKT-PGC-1α signaling pathway. Free Radic. Biol. Med. 2017, 112, 616–630. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lv, L.; Jiang, Z.; Yang, H.; Li, S.; Jiang, Y. Mitofusin 2 protects hepatocyte mitochondrial function from damage induced by GCDCA. PLoS ONE 2013, 8, e65455. [Google Scholar] [CrossRef]
- Yang, X.; Wang, H.; Ni, H.M.; Xiong, A.; Wang, Z.; Sesaki, H.; Ding, W.X.; Yang, L. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 2017, 12, 264–273. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Gao, H.; Lv, Y.; Liu, Y.; Li, J.; Wang, X.; Zhou, Z.; Tipoe, G.L.; Ouyang, S.; Guo, Y.; Zhang, J.; et al. Wolfberry-Derived Zeaxanthin Dipalmitate Attenuates Ethanol-Induced Hepatic Damage. Mol. Nutr. Food Res. 2019, 63, e1801339. [Google Scholar] [CrossRef]
- Luo, J.; Long, Y.; Ren, G.; Zhang, Y.; Chen, J.; Huang, R.; Yang, L. Punicalagin Reversed the Hepatic Injury of Tetrachloromethane by Antioxidation and Enhancement of Autophagy. J. Med. Food 2019, 22, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, Y.; Zhang, S.; Sun, J.; Liu, P.; Xiao, L.; Tang, Y.; Liu, L.; Yao, P. Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy. Nutrients 2016, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Nepal, S.; Kim, M.J.; Lee, E.S.; Kim, J.A.; Choi, D.Y.; Sohn, D.H.; Lee, S.H.; Song, K.; Kim, S.H.; Jeong, G.S.; et al. Modulation of Atg5 expression by globular adiponectin contributes to autophagy flux and suppression of ethanol-induced cell death in liver cells. Food Chem. Toxicol. 2014, 68, 11–22. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Target | Structure | Key Events Regulated by FOXO3a |
|---|---|---|---|
| Melatonin | Sirt3 |  | Increases the expression of SOD2 |
| Punicalagin | Akt |  | Increases antioxidative activities and autophagy |
| gAcrp | AMPK | Induces autophagic process and inhibits apoptosis | |
| Resveratrol | Sirt1 |  | Enhances expression of autophagy-related genes and autophagosome formation |
| Zeaxanthin dipalmitate | P2X7 adipoR1 |  | Modulates PI3k–Akt and AMPK–FOXO3a pathways to restore mitophagy function |
| Quercetin | AMPK ERK2 |  | Activates mitophagy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Zhang, L.; He, J.; Wu, M.; Jia, L.; Guo, J. Role of FOXO3a Transcription Factor in the Regulation of Liver Oxidative Injury. Antioxidants 2022, 11, 2478. https://doi.org/10.3390/antiox11122478
Jin H, Zhang L, He J, Wu M, Jia L, Guo J. Role of FOXO3a Transcription Factor in the Regulation of Liver Oxidative Injury. Antioxidants. 2022; 11(12):2478. https://doi.org/10.3390/antiox11122478
Chicago/Turabian StyleJin, Hong, Li Zhang, Jun He, Min Wu, Li Jia, and Jiabin Guo. 2022. "Role of FOXO3a Transcription Factor in the Regulation of Liver Oxidative Injury" Antioxidants 11, no. 12: 2478. https://doi.org/10.3390/antiox11122478
APA StyleJin, H., Zhang, L., He, J., Wu, M., Jia, L., & Guo, J. (2022). Role of FOXO3a Transcription Factor in the Regulation of Liver Oxidative Injury. Antioxidants, 11(12), 2478. https://doi.org/10.3390/antiox11122478