1. Introduction
The blood–brain barrier (BBB) is a continuous endothelial membrane that insulates the central nervous system from the peripheral immune system to maintain the functional homeostasis of the brain. In response to ischemic stroke, circulating endotoxin levels rise to aggravate brain injury, largely due to the impairment of endothelium integrity [
1,
2]. It is well established that dysregulated systemic immune and inflammatory responses participate in the destruction of the BBB and brain parenchymal damage [
3].
Tight junction (TJ) is a specialized structure that is widely existing in both epithelial cells and endothelial cells, regarded as an important component of BBB. TJ is responsible for restricting the exchange of paracellular and intramembranous components (barrier function), as well as maintaining cell polarity (fence function) [
4]. Zonula occludens-1 (ZO-1) is a scaffold protein that is the first identified as a cytosolic protein in the TJ. By binding to transmembrane proteins, ZO-1 drives the formation of TJ and integrates various signaling pathways, thereby establishing communication between the endothelium and brain [
4]. The loss of ZO-1 and destroyed permeability of the BBB were observed in cerebral ischemic cases associated with oxidative stress and inflammatory responses [
5]. LPS challenge is shown to impair ZO-1 protein in cerebral microvascular endothelial cells [
6]. Inflammation and oxidative stress impair endothelial integrity, but the direct impact on TJ proteins is still unclear.
Oxidative stress is associated with vascular diseases and NADPH oxidases (NOXs) drive reactive oxygen species (ROS) production in the context of inflammation. Different NOX isoforms present in the blood vessel endothelium as the underlying causes of oxidative stress in various heart and cerebrovascular diseases [
7]. NOXs consistently produce low levels of ROS to regulate endothelium-dependent relaxation and redox signaling under physiological conditions, but aberrant function produces excessive ROS leading to vascular injury. Nox2-derived ROS induces endothelial dysfunction associated with alternations in cerebral blood flow [
8]. More pathological factors are involved in brain ischemic injury, but the cause of NOXs activation is remained to be identified. Recently, emerging evidence indicates the potential relation between hypoxia-inducible transcription factor-1
α (HIF-1
α) and NOXs. In colon cancer cells, inhibition of NOX1 expression led to a decrease in the expression of HIF-1
α [
9]. Furthermore, intermittent hypoxia also increased NOX2 expression in the brain cortex of mice, and this effect was abolished in HIF-1
α+/− mice [
10]. HIF-1
α is a transcriptional regulator sensitive to low oxygen tension, but its role in NOXs activation is remained to be elucidated.
Safflower (
Carthamus tinctorius L.) is a kind of traditional Chinese medicine, and its preparation is widely used in the treatment of cardiovascular and cerebrovascular diseases. Hydroxysafflor yellow A (HSYA) is a major active ingredient of safflower, pharmacological studies have confirmed that HSYA improved myocardial ischemia-reperfusion injury through the suppression of NLRP3 inflammasome pathway and TLR4 signaling [
11,
12]. HSYA also inhibited neuronal apoptosis via inhibiting the p38 MAPK signaling pathway [
13]. These events indicate the inhibitory effects of HSYA on inflammation. Furthermore, HSYA is widely used as an antioxidant because it possesses several phenolic hydroxyl groups that donate active hydrogen atoms to interdict oxidative damage (the structure of HSYA was shown in
Figure S1). As medical preparations containing HSYA have been prescribed for the treatment of cardiocerebrovascular diseases, it is necessary to find an integrated mechanism for the better design of therapeutic strategy. In the present study, we identified that inflammation and disturbed redox status induced HIF-1α activation, which drove NOX2 induction in the cerebral microvascular endothelium. HSYA inhibited HIF-1α/NOX2 signaling cascades to protect ZO-1 from proteasomal degradation, resultantly protecting cerebral vessel integrity to attenuate brain injury. Given that conventional antioxidants display poor reactivity with endogenous ROS [
7], targeting ROS-generating enzymes should be a more effective strategy for combating oxidative stress than scavenging highly reactive molecules.
2. Materials and Methods
2.1. Materials and Reagents
HSYA (purity ≥98%) was obtained from Chengdu Must Biotechnology Co, Ltd. (Chengdu, China). LPS (L2880), tert-butyl hydroperoxide (t-BHP, 416665), diamide (D3648), N-acetyl-l-cysteine (NAC, A9165), β-nicotinamide mononucleotide (NMN, N3501), rose bengal (330000), 2,3,5-triphenyltetrazolium chloride (TTC, T8877) and Evans blue (E2129) were purchased from Sigma (St. Louis, MO, USA). PX-478(HY-10231), cycloheximide (HY-12320), and 4-hydroxynonenal (4-HNE, HY-113466) were provided by Med Chem Express (Brea, CA, USA). gp91-ds-tat and sc gp91-ds-tat were obtained from GenScript Co., Ltd. (Nanjing, China).
2.2. Animals and Treatments
The study was approved by the Animal Ethics Committee of China Pharmaceutical University (protocol code: 2020-05-007 and date of approval: 14 May 2020). Male C57BL/6J mice (6–8 weeks old) were purchased from the Experimental Animal Center of Yangzhou University (Yangzhou, China). Mice were housed five per cage with a constant temperature of (24 ± 2 °C) for a 12:12 h light–dark cycle and given free access to standard food and water. They adapted to these conditions for 7 days before being used in the experiments.
For the preparation of the photothrombotic stroke model, male C57BL/6J mice were anesthetized and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). The skull was exposed after incising the midline and removing the periosteum, and then a cold light source (11,500 lux) converged by a 2 mm diameter fiber optic bundle was placed in the position of 1.5 mm to the right of the bregma. After injection of rose bengal (100 mg/kg, i.p.) for 5 min, the indicated position of the skull was illuminated for 15 min. Mice in the sham group underwent the same surgical procedures but received 0.9% saline instead of rose bengal. Following the illumination, the incision was sutured and the mice were transferred for postoperative rehabilitation.
The mice were randomly divided into 4 groups, with 15 mice in each group. After a photothrombotic stroke, mice were intraperitoneally injected with HSYA (50 mg/kg) and NAC (100 mg/kg) at the doses previously published [
12,
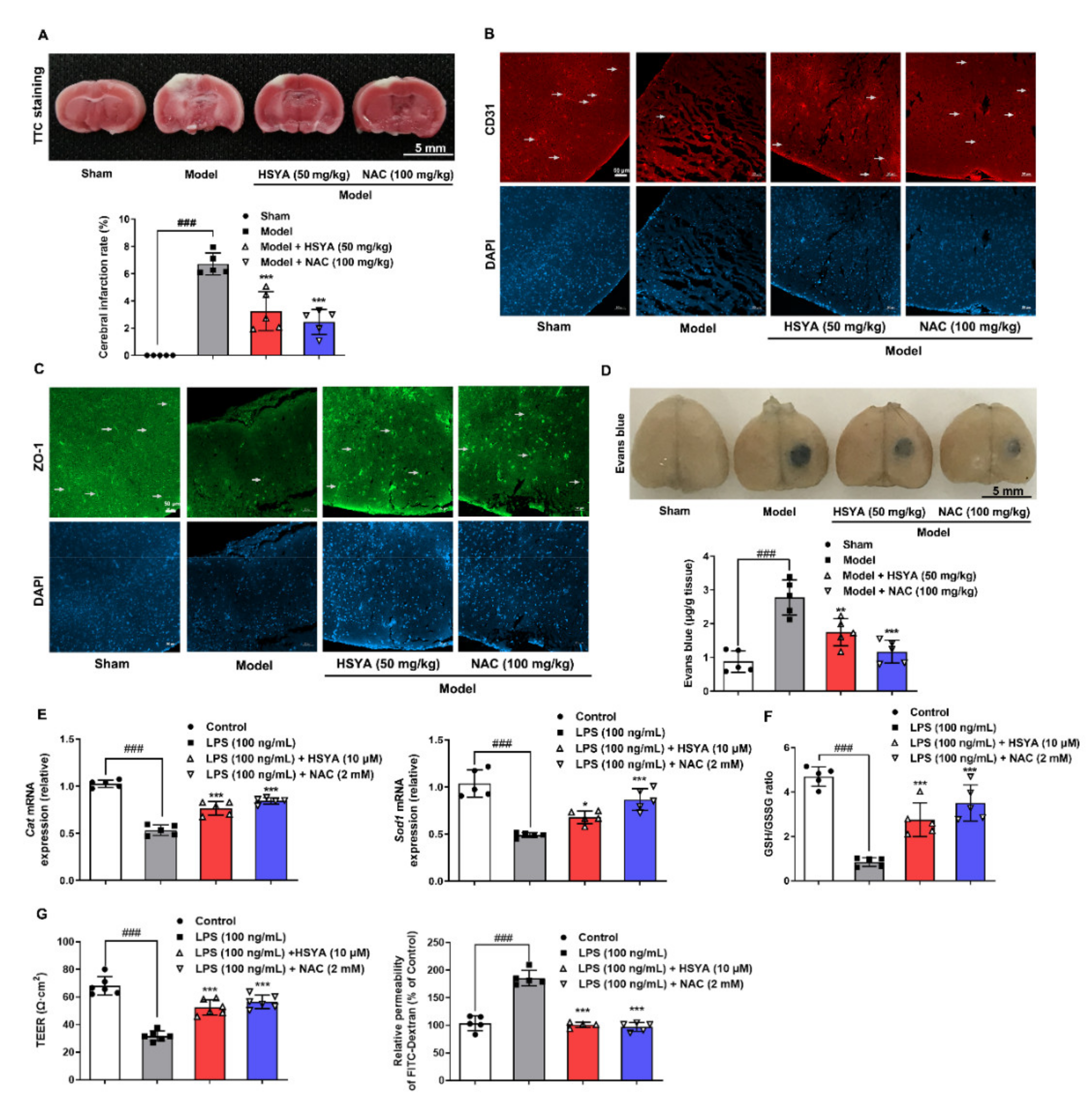
14]. The sham and the model group were given the same volume of normal saline. It was given once a day for 3 days. After administration, the spleen, liver, and kidney of the mice were weighed, and the length of the tibia was measured. The ratios of spleen, liver, and kidney weights to tibia length were calculated. At the same time, 5 mice from each group were randomly selected for TTC staining, Evans blue staining, and immunofluorescence staining of brain tissue.
For TTC staining, the brain of mice was dissected and frozen for 15 min. Afterward, centering on the largest diameter of the infarct area, the brain tissue was cut into coronal slices toward the head, respectively. The slices were stained with TTC (2%, w/v) for 10 min in a 37 °C water bath in the dark and fixed with 4% paraformaldehyde. After 24 h, brain slices were photographed and the infarct area was counted with ImageJ (the cerebral infarction rate = infarct area/total section area × 100%).
2.3. Hematoxylin and Eosin (HE) Staining
The spleen, liver, and kidney tissues were washed with PBS, fixed with 10% formaldehyde, dehydrated, embedded in paraffin, cut into 5 μm sections, and stained with hematoxylin and eosin. The tissue sections were observed under an inverted microscope (OLYMPUS, IXplore Standard, Tokyo, Japan).
2.4. Evans Blue Staining
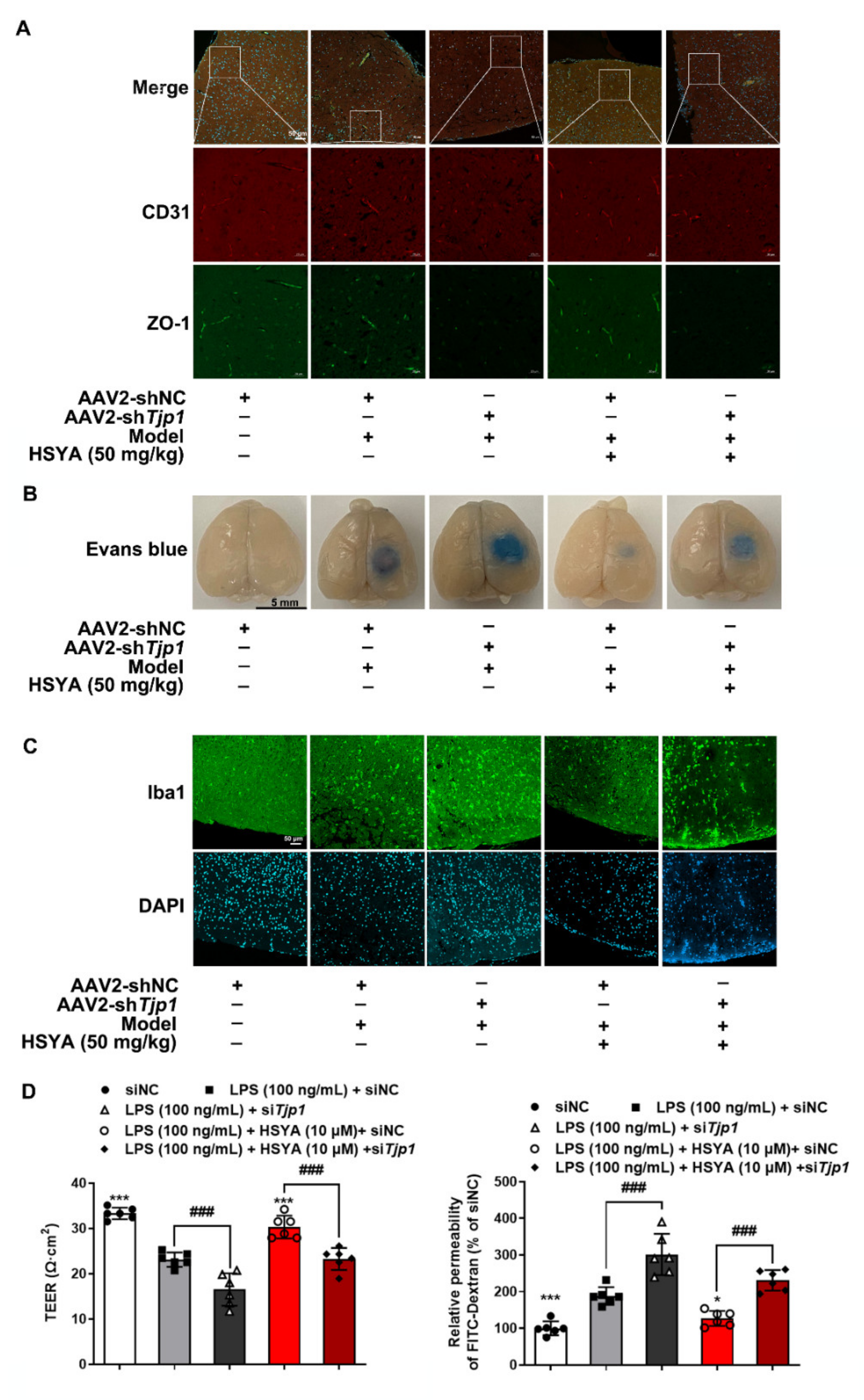
After the administration, mice were injected with 2% Evans blue solution (4 mL/kg body weight) via the tail vein. Two hours after circulation, the mice were perfused with normal saline and paraformaldehyde, in turn, to wash out the residual Evans blue in the blood vessels and fix the tissue. Brain tissues were harvested and photographed, the infarct area was taken and homogenized with 50% trichloroacetic acid. After incubating overnight at 37 °C, tissues were centrifuged at 12,000× g for 20 min. The supernatant was collected and measured absorbance at 620 nm. The content of Evans blue leakage was quantitatively analyzed from the standard curve.
2.5. Cell Culture
bEnd.3 cells from iCell Bioscience Inc (Shanghai, China) were grown in DMEM (KeyGEN BioTECH, Nanjing, China) supplemented with 10% (v/v) FBS (Gibco, New York, NY, USA) at 37 °C in a humidified atmosphere of 5% CO2 in the air.
2.6. RNA Interference
To specifically knockdown Nox1, Nox2, ZO-1, and 20S proteasome, Nox1 small interfering RNA (siRNA), cybb siRNA, Tjp1 siRNA, Psmb9 siRNA, or negative control (NC) siRNA were transfected into bEnd.3 with Lipofectamine™ 3000 transfection reagent (Invitrogen™, L3000015, Carlsbad, CA, USA) when cells grew to a confluence of 70–80%. The siRNA sequences were as follows: siNox1: 5′- CGUCAGCUAUGGAGUUUAU -3′, siCybb: 5′- CACCAUCUCUUUGUGAUCU-3′, siTjp1: 5′-GCGACUAGCUGGUGGAAAU-3′, siPsmb9: 5′-GACUUGUUAGCGCA
UCUCAUA-3′, siNC: 5′-UUCUCCGAACGUGUCACGU-3′ (Shanghai GenePharma Co., Ltd., Shanghai, China). To carry out HIF-1α overexpression, bEnd.3 cells were transfected with pcDNA3.1-M_Hif1α and pcDNA3.1-M_NC plasmids. After culturing for 48 h, bEnd.3 cells were treated with indicated reagents for 16 h.
2.7. Measurement of Lactate Concentration and ELISA Assay
Blood samples collected from the experimental animals were centrifuged at 1000× g for 15 min to obtain serum. IL-1β, LPS, TNF-α in serum were measured by mouse IL-1β ELISA Kit (Neobioscience, Shenzhen, China), mouse LPS ELISA Kit (CUSABIO, Wuhan, China), and mouse TNF-α ELISA Kit (Neobioscience, Shenzhen, China), respectively.
bEnd.3 cells were treated with HSYA (10 μM) or NAC (2 mM) in the presence of LPS (100 ng/mL) for 16 h, referring to the dose in the published literature [
11,
13]. The cell supernatant was collected and lactate concentration was measured according to the manufacturer’s instructions (Jiancheng, A019-2-1, Nanjing, China). Additionally, the treated bEnd.3 cells were washed with PBS and dissociated with trypsin, then the freeze–thaw process was repeated to lyse cells. After centrifuging at 1500×
g for 10 min at 4 °C, the supernatant was collected for the measurement of 4-HNE according to the manufacturer’s protocol of the 4-HNE ELISA Kit (Elabscience, Wuhan, China).
2.8. The Ratio of NAD+/NADH, GSH/GSSG, and the Content of MDA, Protein Carbonyl
bEnd.3 cells were treated with HSYA (10 μM) or NAC (2 mM) in the presence of LPS (100 ng/mL) for 16 h. NAD+/NADH ratio was measured according to the manufacturer’s protocol of NAD+/NADH Quantification Kit (Sigma, MAK037, St. Louis, USA). Briefly, after washing with cold PBS, bEnd.3 were collected, extracted with 400 µL NAD+/NADH extraction buffer, and centrifuged at 13,000× g for 10 min to remove insoluble material. Subsequently, half of the supernatant was collected for the detection of total NADH and NAD (NADtotal), and the remaining half was incubated at 60 °C for 30 min to decompose NAD for the determination of NADH. Then, 10 µL NADH Developer was added for coloration, and the absorbance was measured at 450 nm. NAD+/NADH ratio was obtained according to the standard curve and the formula ratio = (NADtotal-NADH)/NADH.
GSH/GSSG ratio was measured using GSH/GSSG Ratio Detection Assay Kit (Abcam, ab138881, Cambridge, UK). In brief, the treated bEnd.3 cells were washed with PBS and resuspended in Lysis Buffer. The supernatant was collected after centrifuging at 12000× g for 15 min at 4 °C. Deproteinization was carried out with trichloroacetic acid, which was neutralized to pH 4-6 with NaHCO3 later. Finally, fluorescence was measured by a multimode microplate reader (BERTHOLD Technologies, Bad Wildbad, Germany) at excitation of 490 nm and emission of 520 nm.
For measurement of MDA and protein carbonyl, the cell lysis buffer was centrifuged and detected according to the protocols of the manufacturer (Jiancheng, A003-4-1, A087-1-2, Nanjing, China). The absorbance was measured at 370 nm and 530 nm, to calculate the content of MDA and protein carbonyl, respectively.
2.9. Measurement of ROS and Lipid Peroxidation
For the measurement of intracellular ROS generation, the treated bEnd.3 cells were incubated with 5 μM dihydroethidium (DHE, Beyotime, S0063, Shanghai, China) at 37 °C for 30 min in the darkness. After washing with PBS 3 times, the intensity of cytosolic ROS was measured by a confocal laser-scanning microscope (Zeiss, LSM 800, Jena, Germany). For lipid peroxidation level, the treated bEnd.3 cells were incubated with a 10 µM Image-iT Lipid Peroxidation Sensor (Invitrogen, C10445, Carlsbad, CA, USA) for 30 min at 37 °C. Additionally, then nuclear staining was performed using Hoechst (Invitrogen, Carlsbad, CA, USA). Fluorescence images were captured under a confocal laser-scanning microscope (Zeiss, LSM 800, Jena, Germany) at emission 510/590 nm.
To observe ROS production in brain tissues, the brains of experimental animals were frozen and cut into slices. Then, the sections were incubated with 10 μM DCFH-DA (Beyotime, S0033S, Shanghai, China) for 30 min and DAPI for 10 min. The intensity of ROS was measured by a confocal laser-scanning microscope (Zeiss, LSM 800, Jena, Germany).
2.10. Immunofluorescence Staining
To detect the protein expression levels of CD31, Iba1, and ZO-1 in the brains of mice, the brain tissues were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek, Tokyo, Japan) and then cut into 5 μm thick slides. After fixation with 4% paraformaldehyde, the slides were permeabilized with 0.3% Triton X-100 and blocked with goat serum, followed by incubated overnight at 4 °C with primary antibodies (anti-CD31, 1 μg/mL, Abcam Cat# ab9498, RRID: AB_307284; anti-Iba1, 1:100, Abcam Cat# ab178847, RRID: AB_2832244; anti-ZO-1, 1 μg/mL, Abcam Cat# ab216880). After washing for three times, the slides were incubated with second antibody (Goat Anti-Mouse IgG H&L (Alexa Fluor® 647), 1:500, Abcam Cat# ab150115, RRID: AB_2687948; Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488), 1:500, Abcam Cat# ab150077, RRID: AB_2630356; Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 594), 1:500, Abcam Cat# ab150076, RRID: AB_2782993) at room temperature for 2 h. Nuclei were stained with DAPI (1:1000, Bioworld Technology, BD5010, St. Paul, MN, USA) and an anti-fluorescence quenching agent (Beyotime, P0126, Shanghai, China) was added. Slides were observed and captured under a confocal laser-scanning microscope (Zeiss, LSM 800, Jena, Germany).
To detect the cellular immunofluorescence, the treated bEnd.3 cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 5% BSA. After that, cells were incubated with anti-HIF-1α (1:800, Cell Signaling Technology Cat# 36169, RRID: AB_2799095), anti-IgG (1:500, Beyotime Cat# A7016), anti-histone 3 (1:500, Cell Signaling Technology Cat# 4499, RRID:AB_10544537), anti-claudin 5 (1:100, Bioworld Technology Cat# BS1069, RRID: AB_1664057), anti-occludin (1:100, Bioworld Technology Cat# BS72035) or anti-ZO-1 (1 μg/mL, Abcam Cat# ab216880) overnight at 4 °C and goat anti-rabbit IgG H&L (Alexa Fluor® 488) (1:500, Abcam Cat# ab150077, RRID: AB_2630356) at room temperature for 2 h. To achieve the co-localization of p47phox and Nox2, bEnd.3 cells were co-incubated with anti-p47phox (1:100, Santa Cruz Biotechnology Cat# sc-17845, RRID: AB_627986) and anti-Nox2 (1:100, Abcam Cat# ab80508, RRID: AB_1603890) at 4 °C overnight, followed by Goat Anti-Rabbit IgG H&L (Alexa Fluor® 647) (1:500, Abcam Cat# ab150083, RRID: AB_2714032) and Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) (1:500, Abcam Cat# ab150113, RRID: AB_2576208) at room temperature for 2 h. Nuclei were located with DAPI (1:1000, Bioworld Technology, BD5010, St. Paul, MN, USA). Images were captured and analyzed under a confocal laser-scanning microscope (Zeiss, LSM 800, Jena, Germany).
2.11. Western Blotting and Immunoprecipitation
bEnd.3 cells were lysed with RIPA lysis buffer containing phosphatase and protease inhibitors (Roche, Basel, Switzerland) and the protein was quantified with a BCA protein assay kit (Thermo Scientific, 23225, Waltham, MA, USA). An equal amount of sample was separated with SDS-PAGE and then transferred to nitrocellulose membranes. The membranes were blocked in 5% non-fat milk for 2 h and incubated with primary antibodies (anti-Sirt1, 1:1000, Cell Signaling Technology Cat# 9475, RRID: AB_2617130; anti-Von Hippel–Lindau (VHL), 0.03 µg/mL, Abcam Cat# ab77262, RRID: AB_1524559; anti-HIF-1α, 1:1000, Cell Signaling Technology Cat# 36169, RRID: AB_2799095; anti-ZO-1, 1:1000, Abcam Cat# ab216880; anti-claudin 5, 1:1000, Bioworld Technology Cat# BS1069, RRID: AB_1664057; anti-occludin, 1:1000, Bioworld Technology Cat# BS72035; anti-p47phox, 1:500, Santa Cruz Biotechnology Cat# sc-17845, RRID: AB_627986; anti-acetylated-lysine, 1:1000, Cell Signaling Technology Cat# 9441, RRID: AB_331805; anti-proteasome 20S LMP2, 1:10,000, Abcam Cat# ab184172; anti-(Na+, K+)ATPase, 1:1000, Bioworld Technology Cat# BS90909; anti-β-actin, 1:2000, Proteintech Cat# 20536-1-AP, RRID: AB_10700003) at 4 °C overnight. Subsequently, strips were washed with TBST, and co-incubated with corresponding secondary antibodies (Goat Anti-Rabbit IgG (H+L)-HRP, 1:5000, Bioworld Technology Cat# BS13278, RRID: AB_2773728; Rabbit Anti-Goat IgG (H+L)-HRP, 1:5000, Bioworld Technology Cat# BS30503; Goat Anti-Mouse IgG (H + L) HRP, 1:10,000, Bioworld Technology Cat# BS12478, RRID: AB_2773727) at room temperature for 2 h. After further washing, the immunoreactive bands were developed by an ECL kit and then analyzed by Image-Pro Plus 6.0 software.
For immunoprecipitation assay, the lysate was collected and incubated with antibody (anti-Sirt1, 1:30, Abcam Cat# ab189494, RRID: AB_2864311; anti-Nox2, 1:50, Abcam Cat# ab80508, RRID: AB_1603890; anti-VHL, 1:50, Abcam Cat# ab77262, RRID: AB_1524559; anti-ZO-1, 1:100, Abcam Cat# ab216880) overnight at 4 °C and then combined with protein A + G agarose beads (Med Chem Express, HY-K0202, Brea, CA, USA). The mixture was shaken at 4 °C for 4 h to fully combine. The beads were washed with PBS 5 times and boiled with 1% SDS loading buffer for Western blot assay.
Carbonyl introduced into proteins by oxidative reactions was detected with Protein Carbonyl Assay Kit (Western Blot) (Abcam, ab178020, Cambridge, UK). bEnd.3 cells were solubilized with 1× extraction buffer and incubated on ice for 20 min. Protein in cells was treated according to the manufacturer’s protocol. Then, Western blotting was performed to characterize the level of protein carbonyl.
2.12. Quantitative Real-Time PCR (qRT-PCR)
Total RNA from bEnd.3 cells were isolated with an RNA isolator (Vazyme, Nanjing, China). The concentration of RNA was measured by a NANO-100 micro-spectrophotometer (ALLSHENG, China), and the absorbance at 260/280 nm was considered to detect the purity of RNA. HiScript
® Q RT SuperMix for qPCR (Vazyme, Nanjing, China) was used to synthesize cDNA from RNA. Subsequently, qRT-PCR was performed with ChamQ SYBR Color qPCR Master Mix (Vazyme, Nanjing, China) on LightCycler 480 II (Roche, Basel, Switzerland). mRNAs in the tested samples were standardized with
Actb, and the 2
−ΔΔCt method was used for relative quantification. Primer sequences are listed in
Table S1.
2.13. Dual-Luciferase Reporter Gene Assay
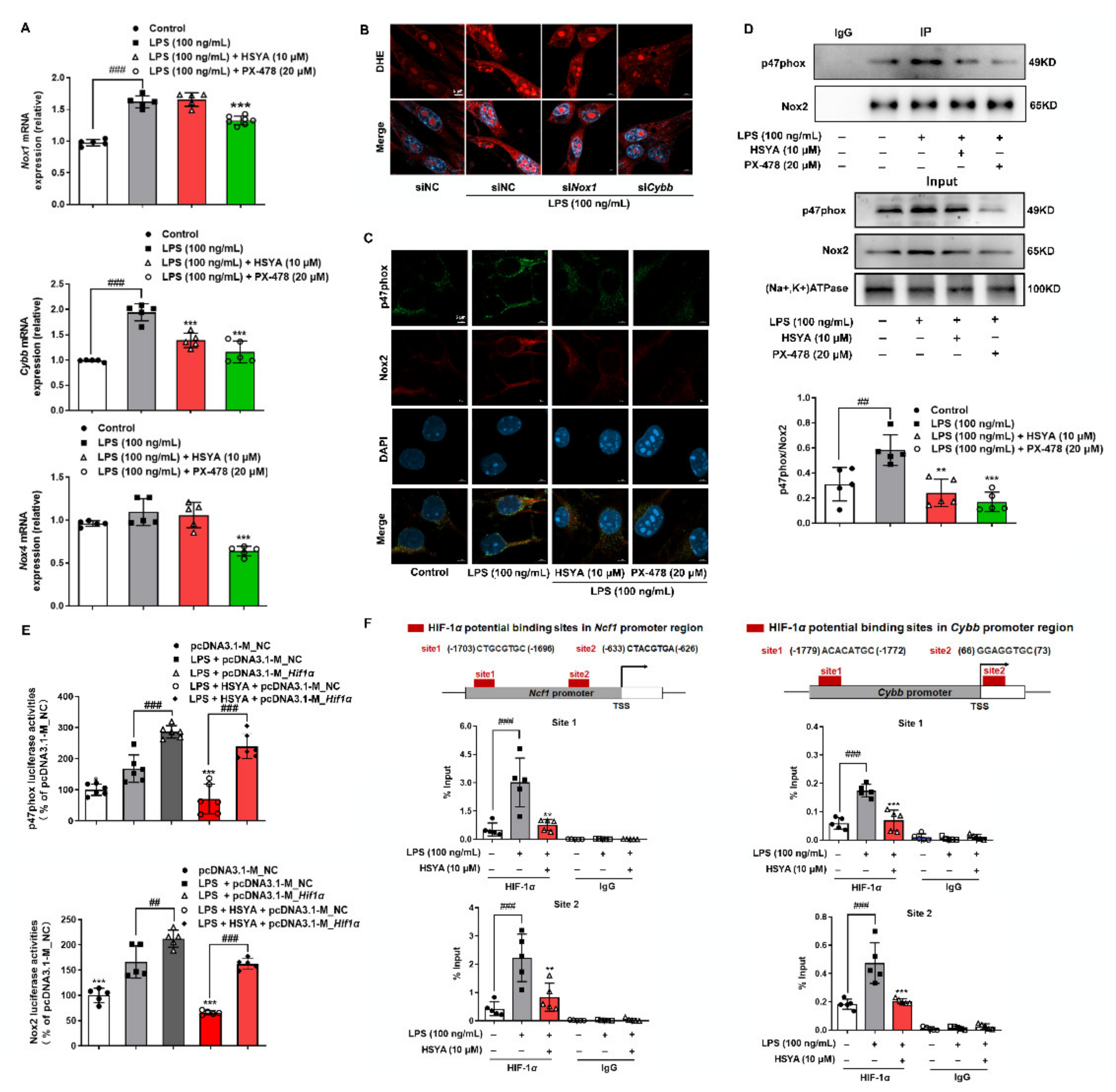
The Mouse_Ncf1 promoter (−2142 to −1) and Mouse_Cybb promoter (−1880 to +74) were synthesized and inserted in the pGL3-basic vector between Mlul and Xhol sites (Genomeditech Co., Ltd., Shanghai, China). bEnd.3 were transfected with pEX3-Ncf1, pEX3-Cybb, pcDNA3.1-M_Hif1α, or pcDNA3.1-M_NC using Lipofectamine™ 3000 reagent and P3000™ Reagent. pRL-SV40 renilla luciferase expression plasmid, a normalized control, were co-transfected into bEnd.3 cells. After 24 h transfection and 16 h stimulation of indicated reagents, bEnd.3 cells were lysed using passive lysis buffer (Promega, E1941, Madison, WI, USA); then, the dual-luciferase assay system (Promega, E2920, Madison, WI, USA) was used to test the luciferase activities.
2.14. Chromatin Immunoprecipitation (ChIP) Analysis
To detect the connection of HIF-1
α with p47phox and Nox2, target chromatin was extracted and precipitated following the manufacturer’s instructions from the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, 9002, Danvers, MA, USA). Briefly, formaldehyde (sigma, 252549, St. Louis, MO, USA) was added to the treated bEnd.3 cells at a final concentration of 1% to make protein cross-linked to DNA. After 10 min, the crosslinking was terminated by the addition of glycine. Cells were washed twice with precooled PBS and collected with PBS buffer containing protease inhibitor cocktail. Chromatin was broken into 150–900 bp fragments by using micrococcal nuclease and sonication. Anti-HIF-1
α was applied for immune precipitation of the target DNA-protein complexes, while normal Rabbit IgG (Cell Signaling Technology, 2729, Danvers, MA, USA) and H3 antibody (Cell Signaling Technology, 4620, Danvers, MA, USA) was used as a negative control and positive control, respectively. An equal part of each sample had been set aside as input control before antibody processing. The bound DNA fragments were eluted and purified and then amplified using loci-specific primer. The primer sequences are shown in
Table S2. Final data were normalized with the Input control of the corresponding sample.
2.15. Adeno-Associated Virus Serotype 2 (AAV2)-Based Endotheliotropic-Specific Knockdown of Tjp1
AAV2/br1-TIE-mir30-m-Tjp1 (NC_000073.7) and AAV2/br1-TIE-NC (negative control) viruses were designed by Hanbio Biotechnology (Shanghai, China). For the endotheliotropic-specific Tjp1 knockdown, mice were injected with 150 μL of AAV2/br1-TIE-mir30-m-Tjp1 at a concentration of 1.5 × 1012 vg/mL viral genomes or AAV2/br1-TIE-NC through the caudal vein. The target sequence of AAV2/br1-TIE-mir30-m-Tjp1 is 5′-GCGACTAGCTGGTGGAAAT-3′, and the sequence of AAV2/br1-TIE-NC is 5′-TTCTCCGAACGTGTCACGT-3′. Three weeks later, the knockout efficiency of ZO-1 was confirmed. Next, the mice were performed with a photothrombotic stroke model and given HSYA (50 mg/kg) for three consecutive days. Mice in the sham group and the model group were given the same volume of normal saline. There were 10 mice in each group, after administration, 5 mice from each group were randomly selected for Evans blue staining to evaluate the BBB function of the knockout mice and immunofluorescence staining in the peri-infarct zones of the brain tissue.
2.16. Transendothelial Electrical Resistance (TEER) Value and FITC–Dextran Paracellular Permeability Determination
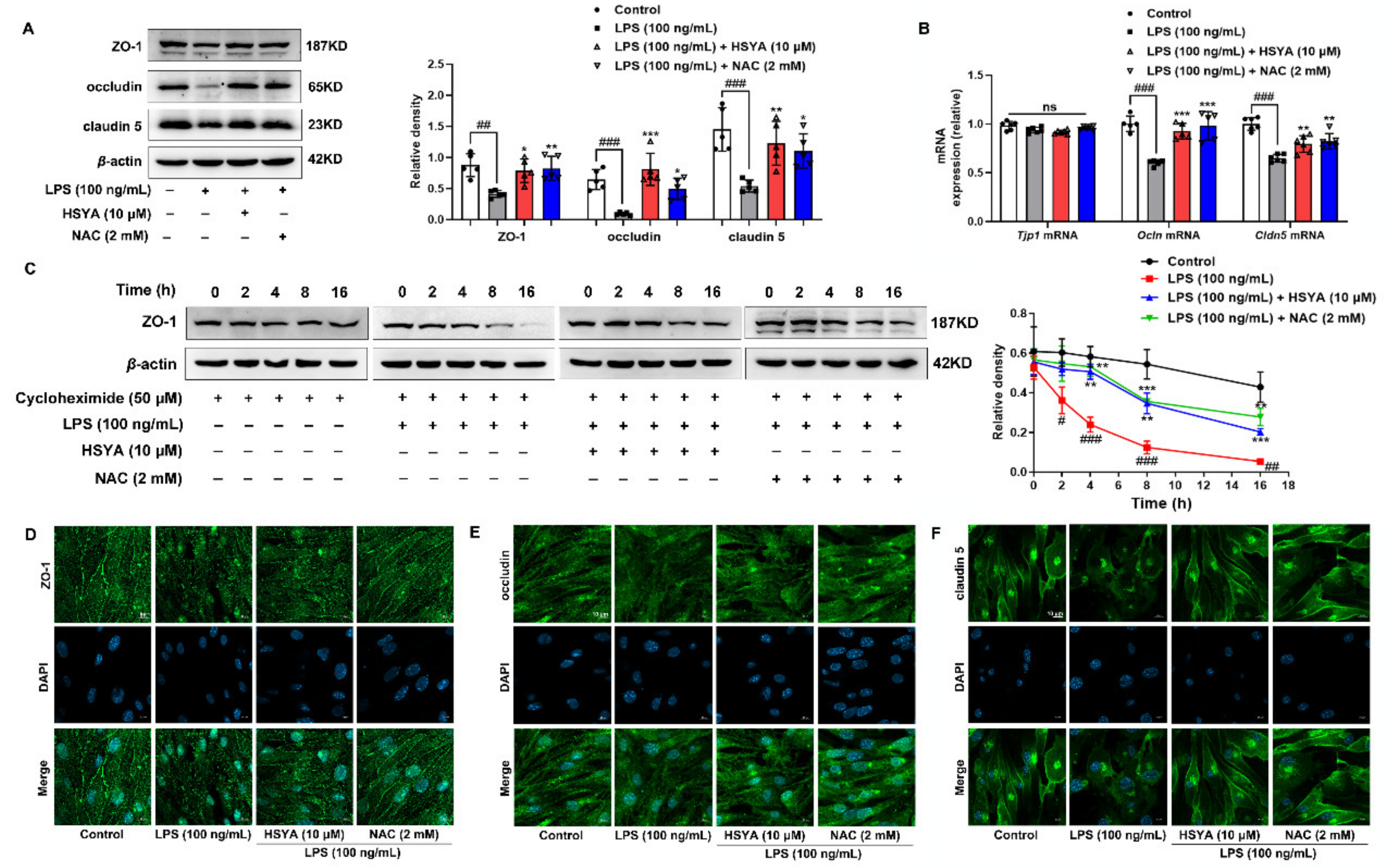
TEER value is generally used to evaluate the integrity of bEnd.3 monolayer cells and the permeability of BBB in vitro. bEnd.3 cells were suspended and seeded in the apical chamber of 12-well Transwell inserts (3460, CORNING, New York, NY, USA) at a volume of 600 μL/well, while 1.5 mL of complete medium was added in the basolateral chamber. After 7 d culture in a sterile incubator, bEnd.3 cells were treated with LPS (100 ng/mL), HSYA (10 μM), and NAC (2 mM) for 16 h. After washing the chambers with HBSS, the TEER value was measured by the R/V Meter of Epithelium (RE1600, Beijing KingTech Technology Co. Ltd., Beijing, China). The TEER values were calculated by subtracting the resistance of a cell-free insert from an insert with cells and by subsequent multiplying by the total membrane surface area to obtain the resistance value in Ω•cm2.
After the measurement of the TEER value, 0.5 mL of DMEM containing 1 mg/mL FITC–Dextran (70 kDa, SIGMA, St. Louis, MO, USA) was added to the apical chamber. Following 1 h incubation in the dark, the fluorescence intensity of FITC–Dextran in the basolateral and apical was measured by a multimode microplate reader (BERTHOLD Technologies, Bad Wildbad, Germany) at excitation 485 nm and emission 525 nm. The equation Pdextran = (RFUbasolateral/RFUapical) (V) (1/time) (1/area) was used to calculate the permeability coefficient, and permeability fold changes were calculated between control and treatment conditions for each experiment.
2.17. Statistical Analysis
All statistical tests are performed by using GraphPad Prism 8.4.2, and the data are expressed as the means ± SD (n ≥ 5). Statistical significance is determined by analysis of ordinary one-way ANOVA, followed by Tukey’s test. All experiments were randomized and blinded to avoid unintentional bias and to generate groups of equal size; values of p < 0.05 were considered statistically significant.
4. Discussion and Conclusions
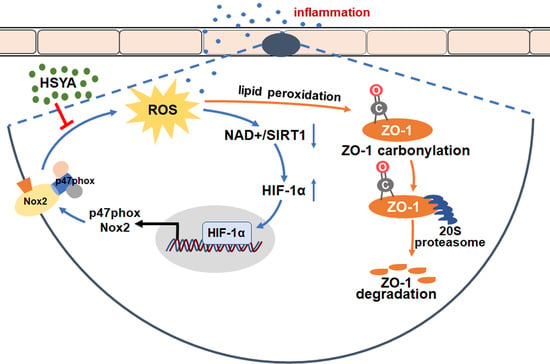
The tight junctions are essential for the integrity of the BBB, wherein ZO-1 stability is a key to the protection of endothelial function. Although there are different isoforms of NOXs in the cerebral microvessels, we identified NOX2 as the predominant isoform that was sensitive to HIF-1α activation. HSYA inhibited HIF-1α/NOX2 signaling cascades to protect ZO-1 from proteasomal degradation, addressing that protection of ZO-1 stability was a way for pharmacological intervention to ensure BBB integrity against ischemic injury.
Cerebral thrombosis is one of the most common types of cerebrovascular disease, and the main cause is atherosclerosis and intima injury, which causes local platelet aggregation and fibrin agglutination, leading to vessel occlusion and consequent ischemic brain injury. Brain injury causes a systemic stress response in the body and activates macrophages in adipose tissue and liver tissue to trigger peripheral inflammatory responses [
21]. In patients with acute stroke, elevated levels of circulating inflammatory cytokines are positively correlated to stroke-associated damage [
22]. We found that circulating levels of inflammatory cytokines including IL-1
β and TNF-
α were significantly upregulated in mice subjected to photothrombotic stroke. Interestingly, the concentration of LPS was also elevated in the periphery blood. By TLR4 activation, LPS evokes oxidative stress and impairs nitric oxide synthase (eNOS) to induce endothelial dysfunction [
23]. It has been reported that BBB permeability and endothelial cell damage are more pronounced in LPS-stimulated brains, accompanied by monocyte infiltration, neuron death, and microglia aggregation [
24]. Consistent with the role in the suppression of neuroinflammation [
25], HSYA reduced proinflammatory cytokines production and combated oxidative stress, having a contribution to preventing the damage to cerebral microvessels.
NOXs also contribute to neuroinflammation and neuronal hyperexcitability in mice after sepsis [
26]. NOXs are an important source of ROS in cardiovascular diseases [
7]. In LPS-activated macrophages, succinate induces HIF-1
α accumulation to evoke inflammation [
27]; however, we found that in microvascular endothelial cells, HIF-1
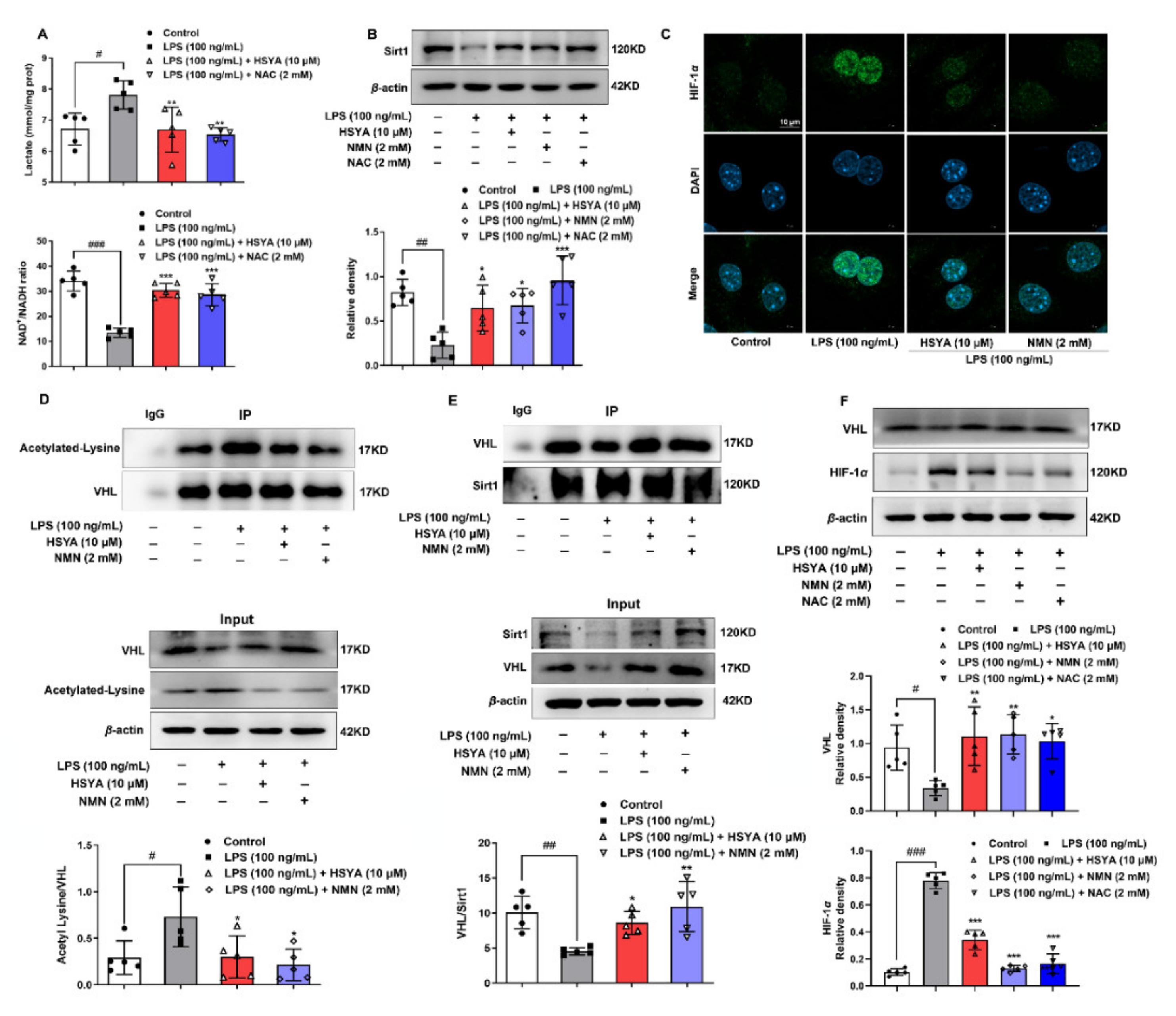
α-mediated NOXs activation was sensitive to altered metabolism and redox state. LPS shifted metabolism to glycolysis, which consumed NAD
+ to inactivate Sirt1 because Sirt1 is an NAD
+-dependent deacetylase. Sirt1 promotes HIF-1
α degradation by VHL induction in skeletal muscle [
17]. By raising NAD
+ contents, HSYA improved Sirt1 activity to prevent HIF-1
α accumulation, largely due to stabilizing VHL through deacetylation modification. These findings are consistent with the published study which demonstrated that Sirt1 induced VHL to promote HIF-1
α degradation efficiently [
17].
HIF-1
α has been studied extensively during hypoxia. HIF-1
α binds to the hypoxia-response elements to regulate gene induction involved in adaptive responses during ischemic injury [
28]. However, accumulating evidence demonstrates the involvement of HIF-1
α-associated inflammation in brain ischemic injury [
29,
30]. Although HIF-1
α was able to increase core catalytic subunits of NOX1 and NOX2, we demonstrated that NOX2 was the predominant isoform in the cerebral microvascular endothelium, and the assembly of cytosolic regulatory subunits with membrane catalytic subunit may be the reason. It has been reported that, in human umbilical vein endothelial cells, NOX2 and HIF-1 were documented to mutually regulate each other to promote angiogenesis [
31]. In the present study, we further revealed that HIF-1
α induction of NOX2 in the context of inflammation and identified p47phox and Nox2 as target genes of HIF-1
α. From the aspect of chemical structure, HSYA could donate reducing equivalents from the phenolic group to scavenge ROS, and its antioxidative effects have been well reported [
11,
25]; however, we addressed that targeting the enzymes that catalyze ROS generation should be a more important means to protect against ischemic injury.
TJ comprises a variety of transmembrane proteins, including claudin and occludin, as well as intracellular scaffold proteins such as ZO-1, -2, and -3 [
5]. ZOs can bind to actin and vinculin-based cytoskeletal filaments, responsible for paracellular permeability in epithelia and endothelia. It was reported that proinflammatory cytokines reduced ZO-1 expression and occludin co-association in human brain microvascular endothelium [
5]. In the present study, we found that ZO-1 protein stability was susceptible to oxidative stress. Accumulated ROS can produce a series of lipid peroxidation products such as 4-HNE and MDA, which can covalently modify proteins for oxidative degradation. The main pathway of protein degradation in mammals is through the proteasome system, including ubiquitin-dependent degradation by 26S proteasome and ubiquitin-independent proteolysis by 20S core proteasome [
32]. The 26S proteasome comprises 20S peptidase and 19S regulatory particles, and actions through ATP-powered protein degradation machinery. Differently, oxidative damage causes protein degradation mainly through the 20S proteasome pathway regardless of ATP. Protein oxidation causes conformational changes, and exposure to hydrophobic residues is recognized by the 20S proteasome, promoting the opening of the
α-rings and the degradation of the
β-rings [
20]. NQO1 could protect C/EBP
α stability from 20S proteasomal degradation in protection against chemical-induced skin cancer [
33]. We demonstrated that the 20S proteasome could bind to the oxidized ZO-1 protein to destroy its stability in the context of inflammation. When the 20S proteasome was knocked down, the protective effect of HSYA on ZO-1 protein was further potentiated. Given the specific impact of ZO-1 deficiency on TEER value and the diffusion of FITC–dextran, it is rational to believe that protecting ZO-1 stability against 20S proteasomal degradation by HSYA has a contribution to improving brain microvessel integrity from the aspect of redox homeostasis.
In mice subjected to a photothrombotic stroke, we also observed that HSYA protected cerebrovascular structure and function and reduced microglia aggregation at the infarct site. These results from the damaged brain provided evidence in vivo to support the findings observed in vitro. We should note that although HSYA stabilization of ZO-1 protected brain microvessel integrity, this was not the only way for its protection in vivo. Cerebral thrombosis is the main cause of stroke. Tissue plasminogen activator (t-PA) causes thrombus fibrinolysis, whereas plasminogen activator inhibitor-1 (PAI-1) leads to thrombus formation by inhibiting t-PA activity. HSYA was shown to improve the outcome following traumatic brain injury by enhancing the t-PA activity and decreasing the PAI-1 activity [
34]. Together with brain microvessel integrity, alleviating neuronal apoptosis and microglial activation by HSYA should have a contribution to cerebral protection [
13,
25]. Furthermore, it has been reported that HSYA possesses vasodilation activity [
35], implicating in antihypertension and pulmonary vascular remodeling [
36]. Therefore, a comprehensive consideration focus on vascular characterization is necessary when we evaluate the protective role of HSYA during cerebral infarction.
In general, we conclude that HIF-1α induction of NOX2 activation underlies microvessel endothelial cell destruction during cerebral infarction, and protection of ZO-1 stability is an important strategy to improve cerebrovascular integrity. HSYA increased ZO-1 expression to rescue cerebrovascular endothelial cells from endotoxin insult by inhibiting HIF-1α/NOX2 signaling cascades, largely due to protecting redox homeostasis. This finding suggests the potential clinical application of HSYA for cerebrovascular protection.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





