
Thioredoxin Reductase-Type Ferredoxin: NADP+ Oxidoreductase of Rhodopseudomonas palustris: Potentiometric Characteristics and Reactions with Nonphysiological Oxidants




Abstract
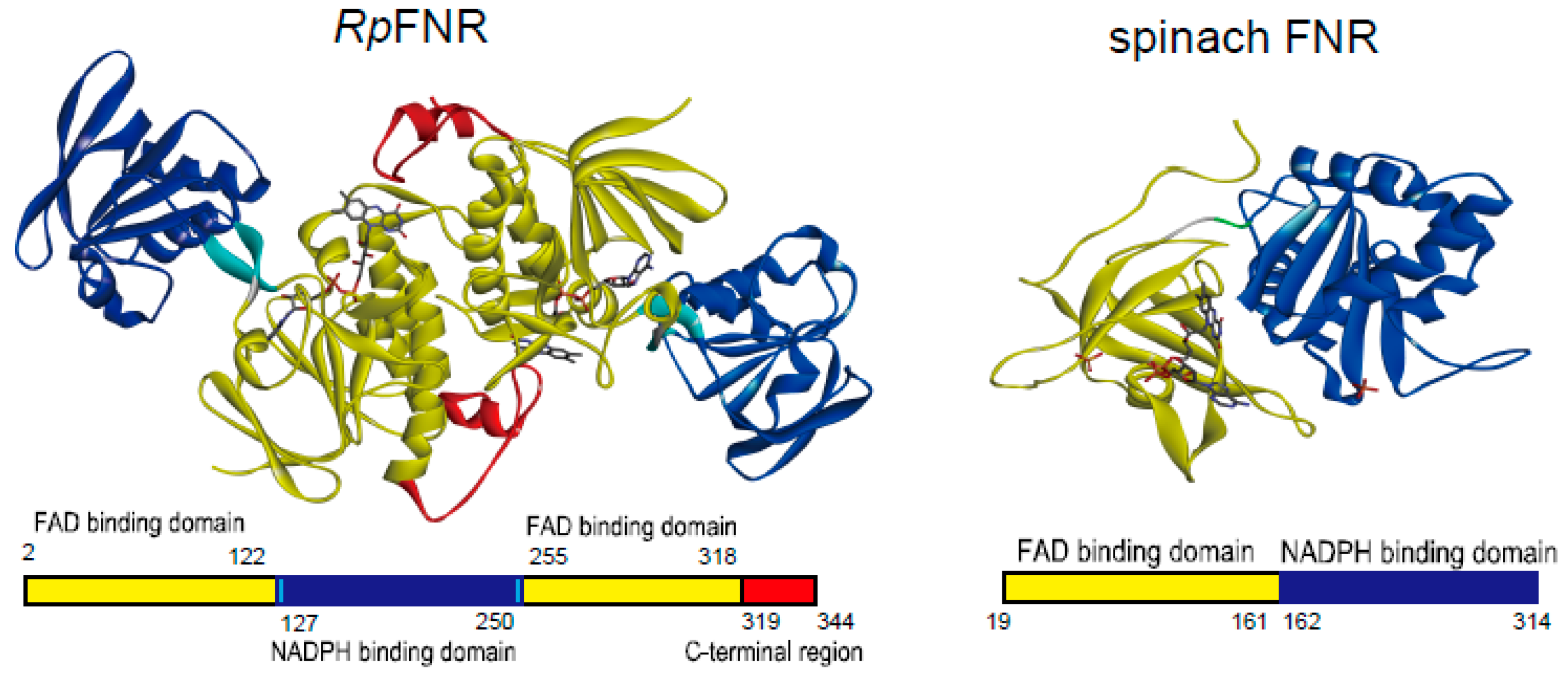
:1. Introduction
2. Materials and Methods
2.1. Enzymes and Reagents
2.2. Photoreduction of RpFNR
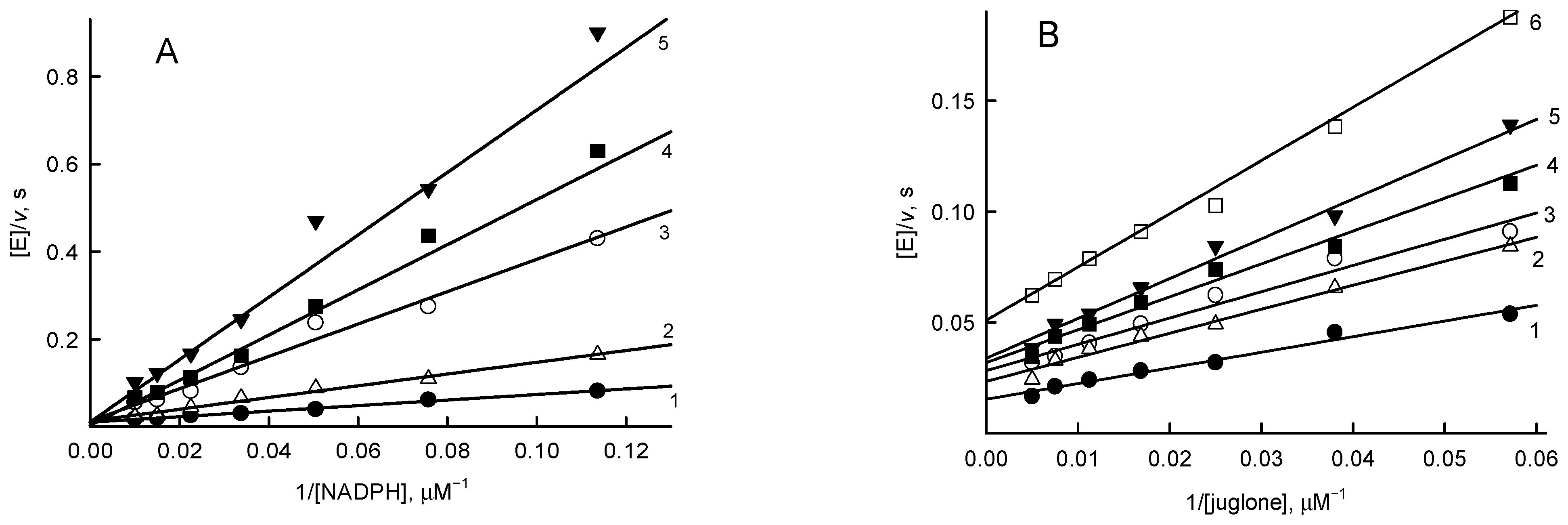
2.3. Steady-State Kinetic Studies
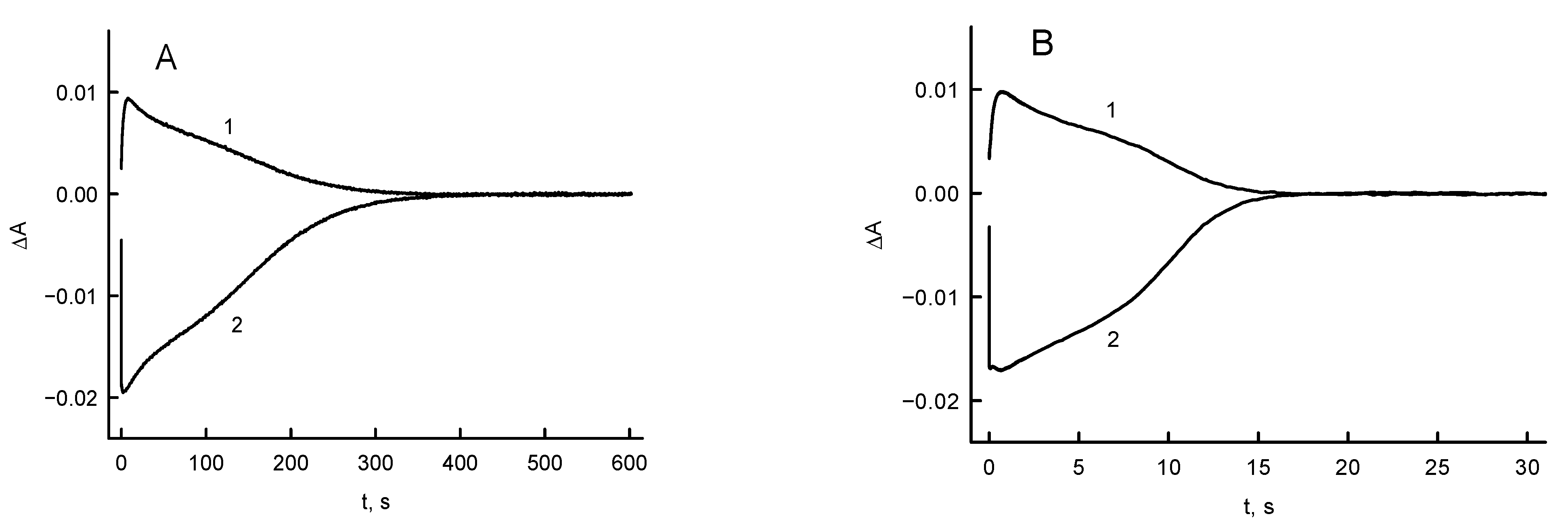
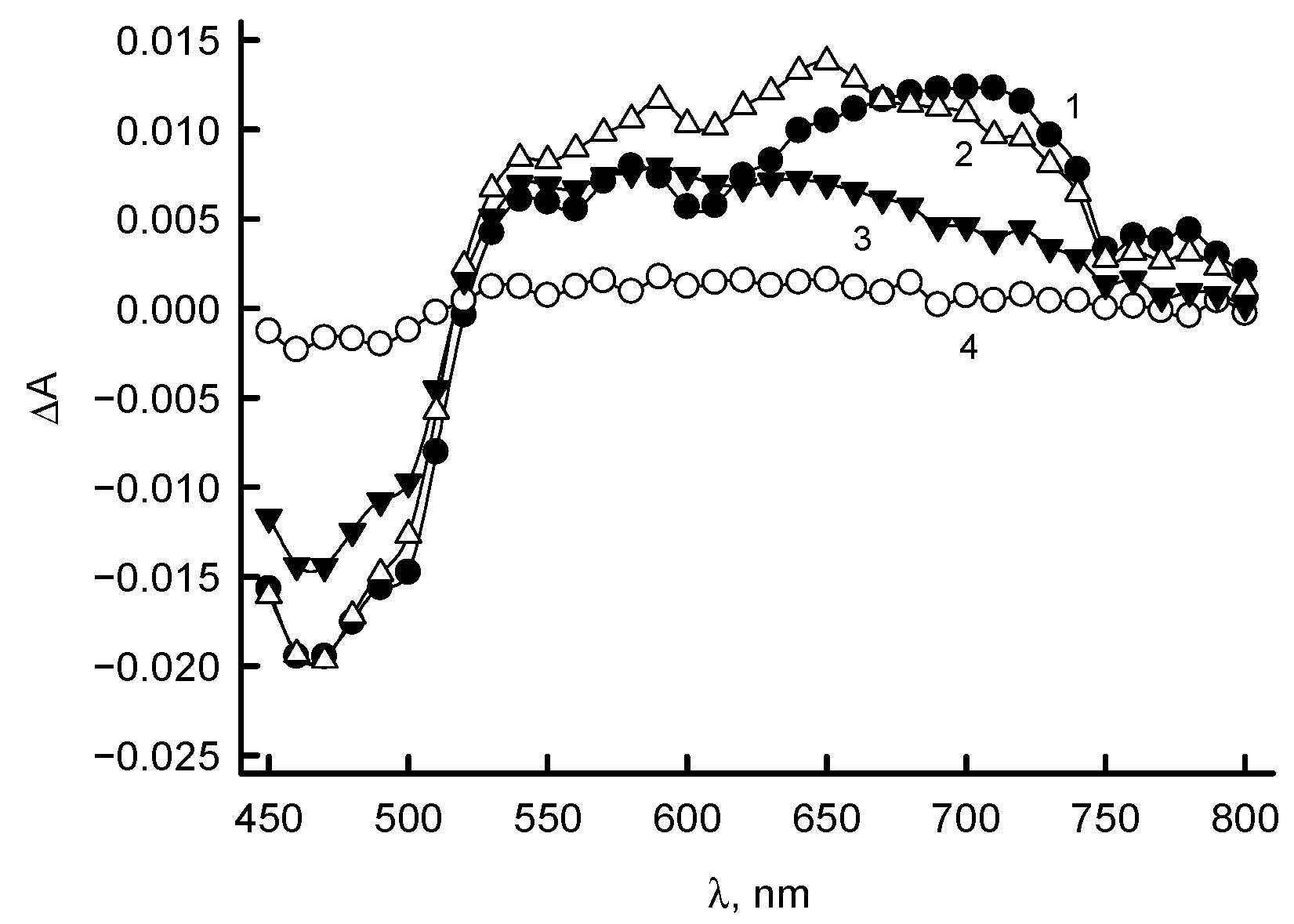
2.4. Presteady-State Kinetic Studies
3. Results
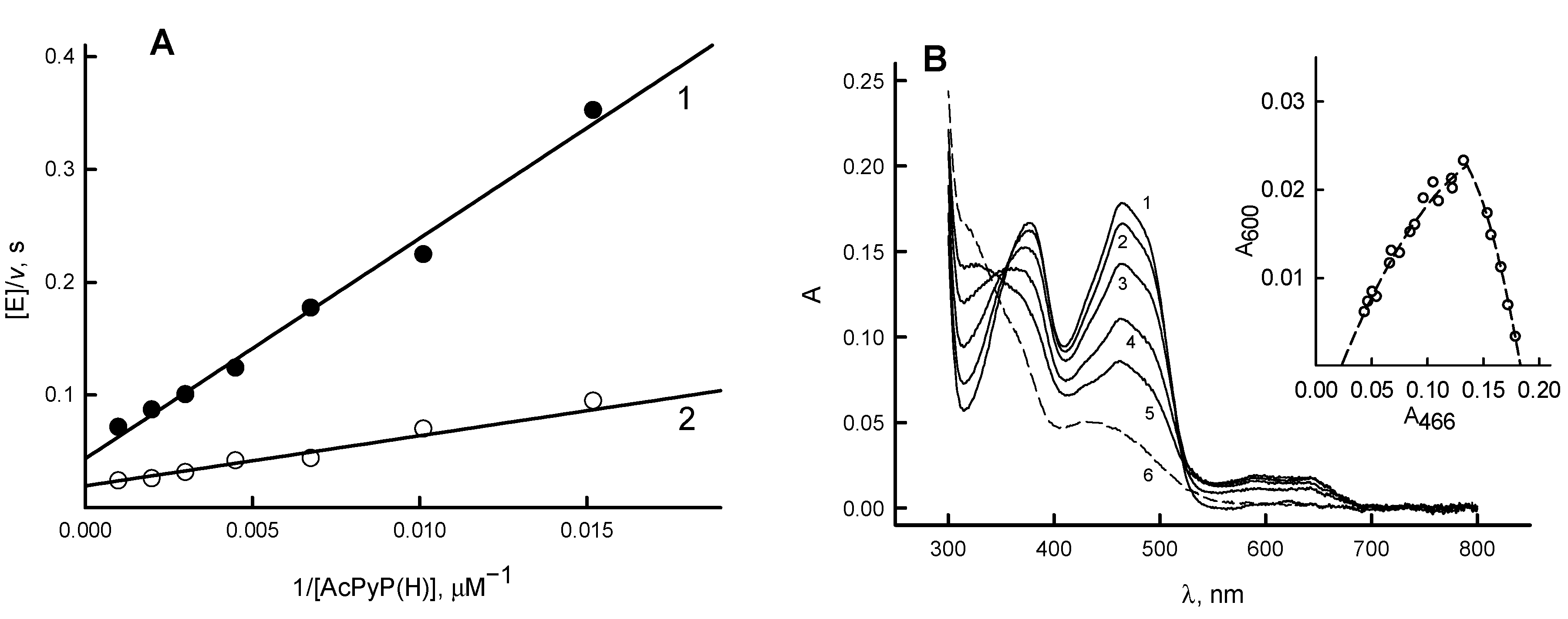
3.1. Determination of Redox Potentials of RpFNR
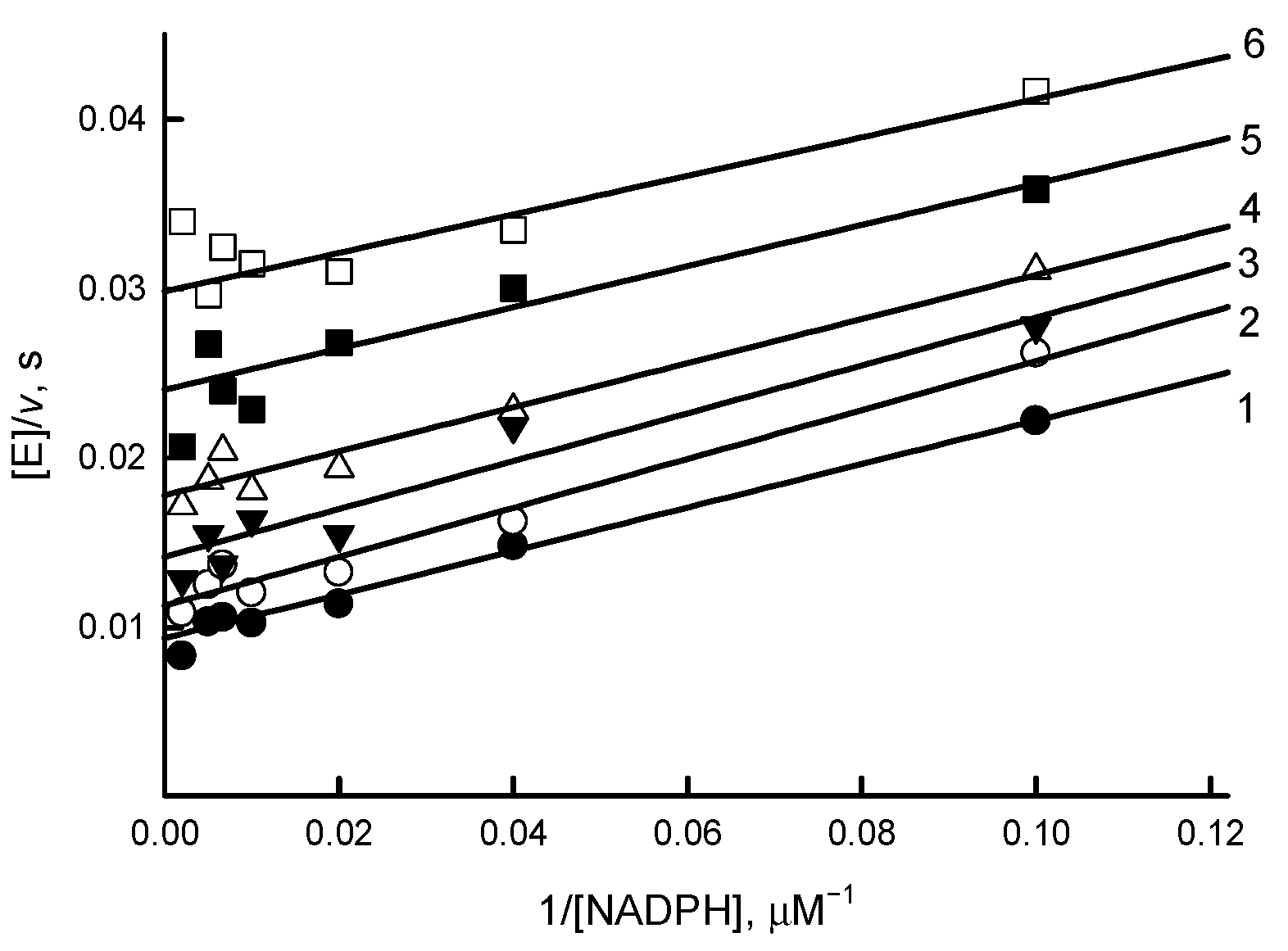
3.2. Steady-State Kinetics and Oxidant Substrate Specificity Studies of RpFNR
3.3. RpFNR Oxidation under Multiple Turnover Conditions
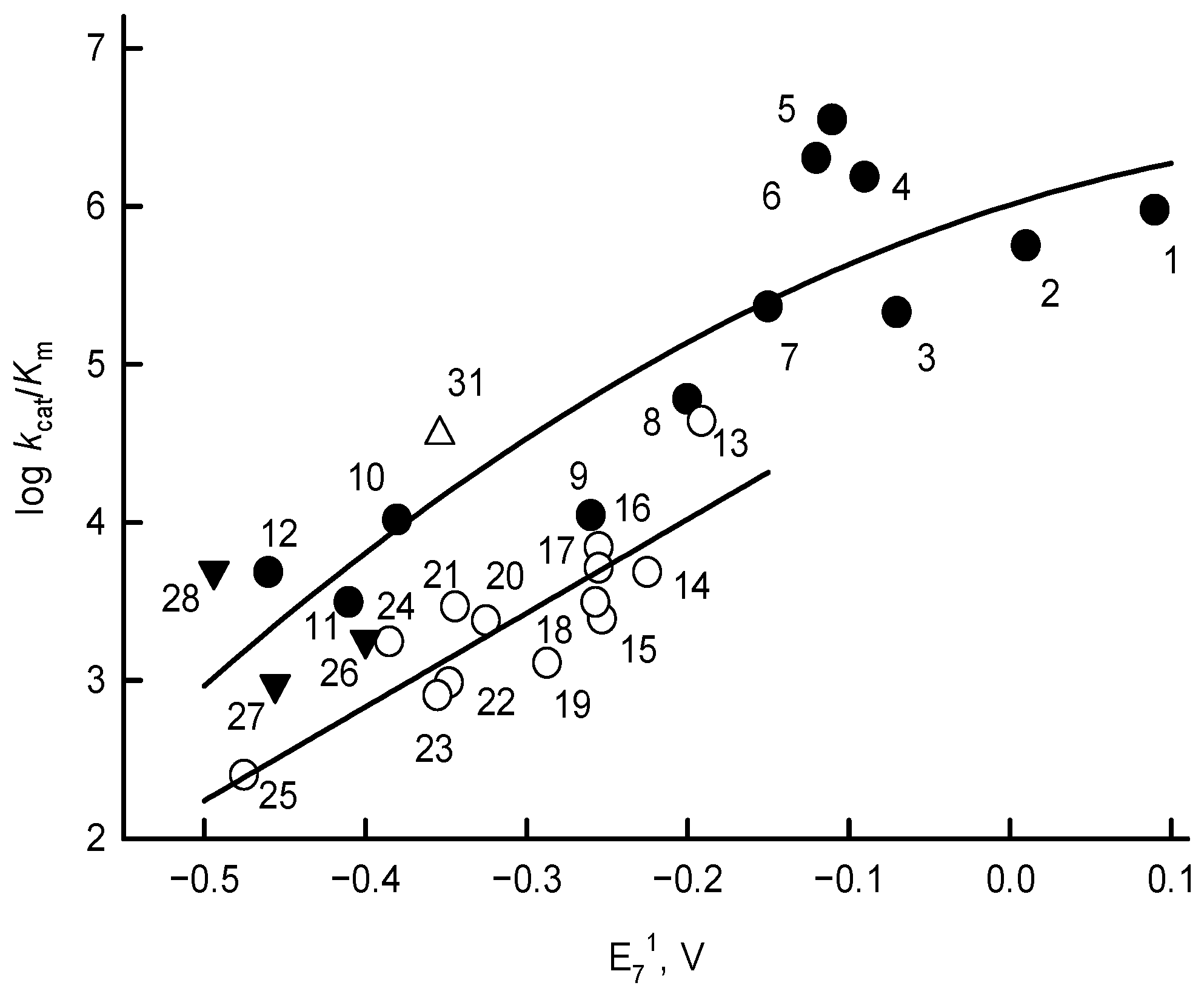
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batie, C.J.; Kamin, H. Electron transfer by ferredoxin: NADP+ reductase. Rapid-reaction evidence for participation of a ternary complex. J. Biol. Chem. 1984, 259, 11976–11985. [Google Scholar] [CrossRef]
- Hurley, J.K.; Morales, R.; Martínez-Júlvez, M.; Brodie, T.B.; Medina, M.; Gómez-Moreno, C.; Tollin, G. Structure–function relationships in Anabaena ferredoxin/ferredoxin: NADP+ reductase electron transfer: Insights from site-directed mutagenesis, transient absorption spectroscopy and X-ray crystallography. Biochim. Biophys. Acta BBA-Bioenerg. 2002, 1554, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, E.A.; Arakaki, A.K.; Cortez, N.; Carrillo, N. Functional plasticity and catalytic efficiency in plant and bacterial ferredoxin-NADP(H) reductases. Biochim. Biophys. Acta BBA-Proteins Proteomics 2004, 1698, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Medina, M. Structural and mechanistic aspects of flavoproteins: Photosynthetic electron transfer from photosystem I to NADP+. FEBS J. 2009, 276, 3942–3958. [Google Scholar] [CrossRef] [PubMed]
- Aliverti, A.; Pandini, V.; Pennati, A.; de Rosa, M.; Zanetti, G. Structural and functional diversity of ferredoxin-NADP+ reductases. Arch. Biochem. Biophys. 2008, 474, 283–291. [Google Scholar] [CrossRef]
- Mulo, P.; Medina, M. Interaction and electron transfer between ferredoxin-NADP+ oxidoreductase and its partners: Structural, functional, and physiological implications. Photosynth. Res. 2017, 134, 265–280. [Google Scholar] [CrossRef]
- Monchietti, P.; López Rivero, A.S.; Ceccarelli, E.A.; Catalano-Dupuy, D.L. A new catalytic mechanism of bacterial ferredoxin-NADP+ reductases due to a particular NADP+ binding mode. Protein Sci. 2021, 30, 2106–2120. [Google Scholar] [CrossRef]
- Corrado, M.E.; Aliverti, A.; Zanetti, G.; Mayhew, S.G. Analysis of the oxidation-reduction potentials of recombinant ferredoxin-NADP+ reductase from spinach chloroplasts. Eur. J. Biochem. 1996, 239, 662–667. [Google Scholar] [CrossRef]
- Kimata-Ariga, Y.; Yuasa, S.; Saitoh, T.; Fukuyama, H.; Hase, T. Plasmodium-specific basic amino acid residues important for the interaction with ferredoxin on the surface of ferredoxin-NADP+ reductase. J. Biochem. 2018, 164, 231–237. [Google Scholar] [CrossRef]
- Hammerstad, M.; Hersleth, H.-P. Overview of structurally homologous flavoprotein oxidoreductases containing the low Mr thioredoxin reductase-like fold—A functionally diverse group. Arch. Biochem. Biophys. 2021, 702, 108826. [Google Scholar] [CrossRef]
- Muraki, N.; Seo, D.; Shiba, T.; Sakurai, T.; Kurisu, G. Asymmetric dimeric structure of ferredoxin-NAD(P)+ oxidoreductase from the green sulfur bacterium Chlorobaculum tepidum: Implications for binding ferredoxin and NADP+. J. Mol. Biol. 2010, 401, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Asano, T. C-terminal residues of ferredoxin-NAD(P)+ reductase from Chlorobaculum tepidum are responsible for reaction dynamics in the hydride transfer and redox equilibria with NADP+/NADPH. Photosynth. Res. 2018, 136, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, H.; Seo, D.; Sakurai, T.; Higuchi, Y. Crystal structure analysis of Bacillus subtilis ferredoxin-NADP+ oxidoreductase and the structural basis for its substrate selectivity. Protein Sci. 2010, 19, 2279–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, D.; Soeta, T.; Sakurai, H.; Sétif, P.; Sakurai, T. Pre-steady-state kinetic studies of redox reactions catalysed by Bacillus subtilis ferredoxin-NADP+ oxidoreductase with NADP+/NADPH and ferredoxin. Biochim. Biophys. Acta BBA-Bioenerg. 2016, 1857, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Okabe, S.; Yanase, M.; Kataoka, K.; Sakurai, T. Studies of interaction of homo-dimeric ferredoxin-NAD(P)+ oxidoreductases of Bacillus subtilis and Rhodopseudomonas palustris, that are closely related to thioredoxin reductases in amino acid sequence, with ferredoxins and pyridine nucleotide coenzymes. Biochim. Biophys. Acta BBA-Proteins Proteomics 2009, 1794, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Seo, D.; Muraki, N.; Kurisu, G. Kinetic and structural insight into a role of the re-face Tyr328 residue of the homodimer type ferredoxin-NADP+ oxidoreductase from Rhodopseudomonas palustris in the reaction with NADP+/NADPH. Biochim. Biophys. Acta BBA-Bioenerg. 2020, 1861, 148140. [Google Scholar] [CrossRef]
- Harwood, C.S.; Gibson, J. Anaerobic and aerobic metabolism of diverse aromatic compounds by the photosynthetic bacterium Rhodopseudomonas palustris. Appl. Environ. Microbiol. 1988, 54, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Donohue, T.J.; Noguera, D.R. Kinetic modeling of anaerobic degradation of plant-derived aromatic mixtures by Rhodopseudomonas palustris. Biodegradation 2021, 32, 179–192. [Google Scholar] [CrossRef]
- Lesanavičius, M.; Aliverti, A.; Šarlauskas, J.; Čėnas, N. Reactions of Plasmodium falciparum ferredoxin: NADP+ oxidoreductase with redox cycling xenobiotics: A mechanistic study. Int. J. Mol. Sci. 2020, 21, 3234. [Google Scholar] [CrossRef]
- Cichocki, B.A.; Donzel, M.; Heimsch, K.C.; Lesanavičius, M.; Feng, L.; Montagut, E.J.; Becker, K.; Aliverti, A.; Elhabiri, M.; Čėnas, N.; et al. Plasmodium falciparum ferredoxin-NADP+ reductase-catalyzed redox cycling of plasmodione generates both predicted key drug metabolites: Implication for antimalarial drug development. ACS Infect. Dis. 2021, 7, 1996–2012. [Google Scholar] [CrossRef]
- Čėnas, N.; Nemeikaitė-Čėnienė, A.; Sergedienė, E.; Nivinskas, H.; Anusevičius, Ž.; Šarlauskas, J. Quantitative structure—Activity relationships in enzymatic single-electron reduction of nitroaromatic explosives: Implications for their cytotoxicity. Biochim. Biophys. Acta BBA-Gen. Subj. 2001, 1528, 31–38. [Google Scholar] [CrossRef]
- Hay, M.P.; Gamage, S.A.; Kovacs, M.S.; Pruijn, F.B.; Anderson, R.F.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. Structure-activity relationships of 1,2,4-benzotriazine 1,4-dioxides as hypoxia-selective analogues of tirapazamine. J. Med. Chem. 2003, 46, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ross, W.C. Tumour-growth inhibitory nitrophenylaziridines and related compounds: Structure-activity relationships. Chem. Biol. Interact. 1969, 1, 27–47. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Tollin, G. Semiquinone formation in flavo- and metalloflavoproteins. Radic. Biochem. 1983, 108, 109–138. [Google Scholar] [CrossRef]
- Mayhew, S.G. The effects of pH and semiquinone formation on the oxidation-reduction potentials of flavin mononucleotide. A reappraisal. Eur. J. Biochem. 1999, 265, 698–702. [Google Scholar] [CrossRef]
- Kaplan, N.O.; Ciotti, M.M. Chemistry and properties of the 3-acetylpyridine analogue of diphosphopyridine nucleotide. J. Biol. Chem. 1956, 221, 823–832. [Google Scholar] [CrossRef]
- Chance, B. A simple relationship for a calculation of the “on” velocity constant in enzyme reactions. Arch. Biochem. Biophys. 1957, 71, 130–136. [Google Scholar] [CrossRef]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Čėnas, N.; Nemeikaitė-Čėnienė, A.; Kosychova, L. Single- and two-electron reduction of nitroaromatic compounds by flavoenzymes: Mechanisms and implications for cytotoxicity. Int. J. Mol. Sci. 2021, 22, 8534. [Google Scholar] [CrossRef]
- Iyanagi, T.; Yamazaki, I. One-electron-transfer reactions in biochemical systems V. Difference in the mechanism of quinone reduction by the NADH dehydrogenase and the NAD(P)H dehydrogenase (DT-diaphorase). Biochim. Biophys. Acta BBA-Bioenerg. 1970, 216, 282–294. [Google Scholar] [CrossRef]
- Anusevičius, Ž.; Martínez-Júlvez, M.; Genzor, C.G.; Nivinskas, H.; Gómez-Moreno, C.; Čėnas, N. Electron transfer reactions of Anabaena PCC 7119 ferredoxin:NADP+ reductase with nonphysiological oxidants. Biochim. Biophys. Acta BBA-Bioenerg. 1997, 1320, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Balconi, E.; Pennati, A.; Crobu, D.; Pandini, V.; Cerutti, R.; Zanetti, G.; Aliverti, A. The ferredoxin-NADP+ reductase/ferredoxin electron transfer system of Plasmodium falciparum. FEBS J. 2009, 276, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Faro, M.; Gómez-Moreno, C.; Stankovich, M.; Medina, M. Role of critical charged residues in reduction potential modulation of ferredoxin-NADP+ reductase. Eur. J. Biochem. 2002, 269, 2656–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M.; Martínez-Júlvez, M.; Hurley, J.K.; Tollin, G.; Gómez-Moreno, C. Involvement of Glutamic Acid 301 in the Catalytic Mechanism of Ferredoxin-NADP+ Reductase from Anabaena PCC 7119. Biochemistry 1998, 37, 2715–2728. [Google Scholar] [CrossRef] [PubMed]
- Aliverti, A.; Deng, Z.; Ravasi, D.; Piubelli, L.; Karplus, P.A.; Zanetti, G. Probing the function of the invariant glutamyl residue 312 in spinach ferredoxin-NADP+ reductase. J. Biol. Chem. 1998, 273, 34008–34015. [Google Scholar] [CrossRef] [Green Version]
- Swenson, R.P.; Krey, G.D. Site-directed mutagenesis of tyrosine-98 in the flavodoxin from Desulfovibrio vulgaris (Hildenborough): Regulation of oxidation-reduction properties of the bound FMN cofactor by aromatic, solvent, and electrostatic interactions. Biochemistry 1994, 33, 8505–8514. [Google Scholar] [CrossRef]
- Nogués, I.; Tejero, J.; Hurley, J.K.; Paladini, D.; Frago, S.; Tollin, G.; Mayhew, S.G.; Gómez-Moreno, C.; Ceccarelli, E.A.; Carrillo, N.; et al. Role of the C-terminal tyrosine of ferredoxin-nicotinamide adenine dinucleotide phosphate reductase in the electron transfer processes with its protein partners ferredoxin and flavodoxin. Biochemistry 2004, 43, 6127–6137. [Google Scholar] [CrossRef]
- Bradley, L.H.; Swenson, R.P. Role of hydrogen bonding interactions to N(3)H of the flavin mononucleotide cofactor in the modulation of the redox potentials of the Clostridium beijerinckii flavodoxin. Biochemistry 2001, 40, 8686–8695. [Google Scholar] [CrossRef]
- Ziegler, G.A.; Vonrhein, C.; Hanukoglu, I.; Schulz, G.E. The structure of adrenodoxin reductase of mitochondrial P450 systems: Electron transfer for steroid biosynthesis. J. Mol. Biol. 1999, 289, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta BBA-Rev. Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Wardman, P.; Dennis, M.F.; Everett, S.A.; Patel, K.B.; Stratford, M.R.L.; Tracy, M. Radicals from one-electron reduction of nitro compounds, aromatic N-oxides and quinones: The kinetic basis for hypoxia-selective, bioreductive drugs. Biochem. Soc. Symp. 1995, 61, 171–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeikaitė-Čėnienė, A.; Šarlauskas, J.; Jonušienė, V.; Marozienė, A.; Misevičienė, L.; Yantsevich, A.V.; Čėnas, N. Kinetics of flavoenzyme-catalyzed reduction of tirapazamine derivatives: Implications for their prooxidant cytotoxicity. Int. J. Mol. Sci. 2019, 20, 4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anusevičius, Ž.; Misevičienė, L.; Medina, M.; Martinez-Julvez, M.; Gomez-Moreno, C.; Čėnas, N. FAD semiquinone stability regulates single- and two-electron reduction of quinones by Anabaena PCC7119 ferredoxin:NADP+ reductase and its Glu301Ala mutant. Arch. Biochem. Biophys. 2005, 437, 144–150. [Google Scholar] [CrossRef]
- Sánchez-Azqueta, A.; Musumeci, M.A.; Martínez-Júlvez, M.; Ceccarelli, E.A.; Medina, M. Structural backgrounds for the formation of a catalytically competent complex with NADP(H) during hydride transfer in ferredoxin-NADP+ reductases. Biochim. Biophys. Acta BBA-Bioenerg. 2012, 1817, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, J.; Peregrina, J.R.; Martínez-Júlvez, M.; Gutiérrez, A.; Gómez-Moreno, C.; Scrutton, N.S.; Medina, M. Catalytic mechanism of hydride transfer between NADP+/H and ferredoxin-NADP+ reductase from Anabaena PCC 7119. Arch. Biochem. Biophys. 2007, 459, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, T.; Sakamoto, H.; Sugiyama, T.; Yamano, T. Formation of the semiquinone form in the anaerobic reduction of adrenodoxin reductase by NADPH. Resonance Raman, EPR, and optical spectroscopic evidence. J. Biol. Chem. 1982, 257, 12075–12080. [Google Scholar] [CrossRef]
- Sakamoto, H.; Ohta, M.; Miura, R.; Sugiyama, T.; Yamano, T.; Miyake, Y. Studies on the reaction mechanism of NADPH-adrenodoxin reductase with NADPH. J. Biochem. 1982, 92, 1941–1950. [Google Scholar] [CrossRef]
- Seo, D.; Asano, T.; Komori, H.; Sakurai, T. Role of the C-terminal extension stacked on the re-face of the isoalloxazine ring moiety of the flavin adenine dinucleotide prosthetic group in ferredoxin-NADP+ oxidoreductase from Bacillus subtilis. Plant Physiol. Biochem. 2014, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Mauk, A.G.; Scott, R.A.; Gray, H.B. Distances of electron transfer to and from metalloprotein redox sites in reactions with inorganic complexes. J. Am. Chem. Soc. 1980, 102, 4360–4363. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | E17 (V) | kcat(app) (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| Quinones | ||||
| 1 | 1,4-Benzoquinone | 0.090 | 130 ± 16 | 9.4 ± 0.8 × 105 |
| 2 | 2-CH3-1,4-benzoquinone | 0.010 | 130 ± 12 | 5.6 ± 0.6 × 105 |
| 3 | 2,6-(CH3)2-1,4-benzoquinone | −0.070 | 52.1 ± 1.8 | 2.1 ± 0.13 × 105 |
| 4 | 5-OH-1,4-naphthoquinone | −0.090 | 138.5 ± 9.3 | 1.5 ± 0.23 × 106 |
| 5 | 5,8-(OH)2-1,4-naphthoquinone | −0.110 | 45.4 ± 3.4 | 3.5 ± 0.2 × 106 |
| 6 | 9,10-Phenanthrene quinone | −0.120 | 34.6 ± 2.4 | 2.0 ± 0.4 × 106 |
| 7 | 1,4-Naphthoquinone | −0.150 | 110 ± 13 | 2.3 ± 0.4 × 105 |
| 8 | 2-CH3-1,4-naphthoquinone | −0.200 | 21.6 ± 2.1 | 6.0 ± 0.8 × 104 |
| 9 | (CH3)4-1,4-benzoquinone(duroquinone) | −0.260 | 4.07 ± 0.53 | 1.1 ± 0.1 × 104 |
| 10 | 9,10-Anthraquinone-2-sulphonate | −0.380 | 3.56 ± 0.33 | 1.0 ± 0.16 × 104 |
| 11 | 2-OH-1,4-naphthoquinone | −0.410 | 0.26 ± 0.03 | 3.1 ± 0.2 × 103 |
| 12 | 2-CH3-3-OH-1,4-naphthoquinone | −0.460 | 1.35 ± 0.13 | 4.8 ± 0.4 × 103 |
| Nitroaromatic compounds | ||||
| 13 | Tetryl | −0.191 | 5.69 ± 0.14 | 4.35 ± 0.30 × 104 |
| 14 | N-methylpicramide | −0.225 | 1.93 ± 0.26 | 4.8 ± 0.6 × 103 |
| 15 | 2,4,6-Trinitrotoluene | −0.253 | 1.30 ± 0.13 | 2.43 ± 0.14 × 103 |
| 16 | Nifuroxime | −0.255 | 4.40 ± 0.32 | 6.9 ± 0.4 × 103 |
| 17 | Nitrofurantoin | −0.255 | 2.21 ± 0.12 | 5.1 ± 0.5 × 103 |
| 18 | p-Dinitrobenzene | −0.257 | 2.21 ± 0.35 | 3.1 ± 0.2 × 103 |
| 19 | o-Dinitrobenzene | −0.287 | 0.48 ± 0.07 | 1.28 ± 0.2 × 103 |
| 20 | 4-Nitrobenzaldehyde | −0.325 | 0.97 ± 0.13 | 2.38 ± 0.4 × 103 |
| 21 | 3,5-Dinitrobenzoic acid | −0.344 | 0.09 ± 0.01 | 2.91 ± 0.2 × 103 |
| 22 | m-Dinitrobenzene | −0.348 | 0.42 ± 0.06 | 9.6 ± 0.7 × 102 |
| 23 | 4-Nitroacetophenone | −0.355 | 0.30 ± 0.05 | 8.0 ± 0.67 × 102 |
| 24 | CB-1954 | −0.385 | 0.52 ± 0.05 | 1.75 ± 0.14 × 103 |
| 25 | 4-Nitrobenzyl alcohol | −0.475 | 0.23 ± 0.03 | 2.50 ± 0.16 × 102 |
| Aromatic N-oxides | ||||
| 26 | 7-F-tirapazamine | −0.400 | 1.20 ± 0.11 | 1.80 ± 0.31 × 103 |
| 27 | Tirapazamine | −0.456 | 0.53 ± 0.04 | 9.41 ± 0.82 × 102 |
| 28 | 7-C2H5O-tirapazamine | −0.494 | 0.46 ± 0.03 | 4.91 ± 0.32 × 103 |
| Single-electron acceptors | ||||
| 29 | Ferricyanide a | 0.410 | 394 ± 19 | 8.8 ± 1.0 × 106 |
| 30 | Fe(EDTA)− | 0.120 | 1.2 ± 0.1 | 2.4 ± 0.2 × 103 |
| 31 | Benzylviologen | −0.354 | 19.6 ± 2.3 | 3.6 ± 0.3 × 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lesanavičius, M.; Seo, D.; Čėnas, N. Thioredoxin Reductase-Type Ferredoxin: NADP+ Oxidoreductase of Rhodopseudomonas palustris: Potentiometric Characteristics and Reactions with Nonphysiological Oxidants. Antioxidants 2022, 11, 1000. https://doi.org/10.3390/antiox11051000
Lesanavičius M, Seo D, Čėnas N. Thioredoxin Reductase-Type Ferredoxin: NADP+ Oxidoreductase of Rhodopseudomonas palustris: Potentiometric Characteristics and Reactions with Nonphysiological Oxidants. Antioxidants. 2022; 11(5):1000. https://doi.org/10.3390/antiox11051000
Chicago/Turabian StyleLesanavičius, Mindaugas, Daisuke Seo, and Narimantas Čėnas. 2022. "Thioredoxin Reductase-Type Ferredoxin: NADP+ Oxidoreductase of Rhodopseudomonas palustris: Potentiometric Characteristics and Reactions with Nonphysiological Oxidants" Antioxidants 11, no. 5: 1000. https://doi.org/10.3390/antiox11051000
APA StyleLesanavičius, M., Seo, D., & Čėnas, N. (2022). Thioredoxin Reductase-Type Ferredoxin: NADP+ Oxidoreductase of Rhodopseudomonas palustris: Potentiometric Characteristics and Reactions with Nonphysiological Oxidants. Antioxidants, 11(5), 1000. https://doi.org/10.3390/antiox11051000