
Metabolic Shades of S-D-Lactoylglutathione




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Chemistry, Preparation and Biochemistry of S-D-Lactoylglutathione and Its Measurement in Biological Samples
2.1. Chemistry
2.2. Preparation
2.3. Biochemistry
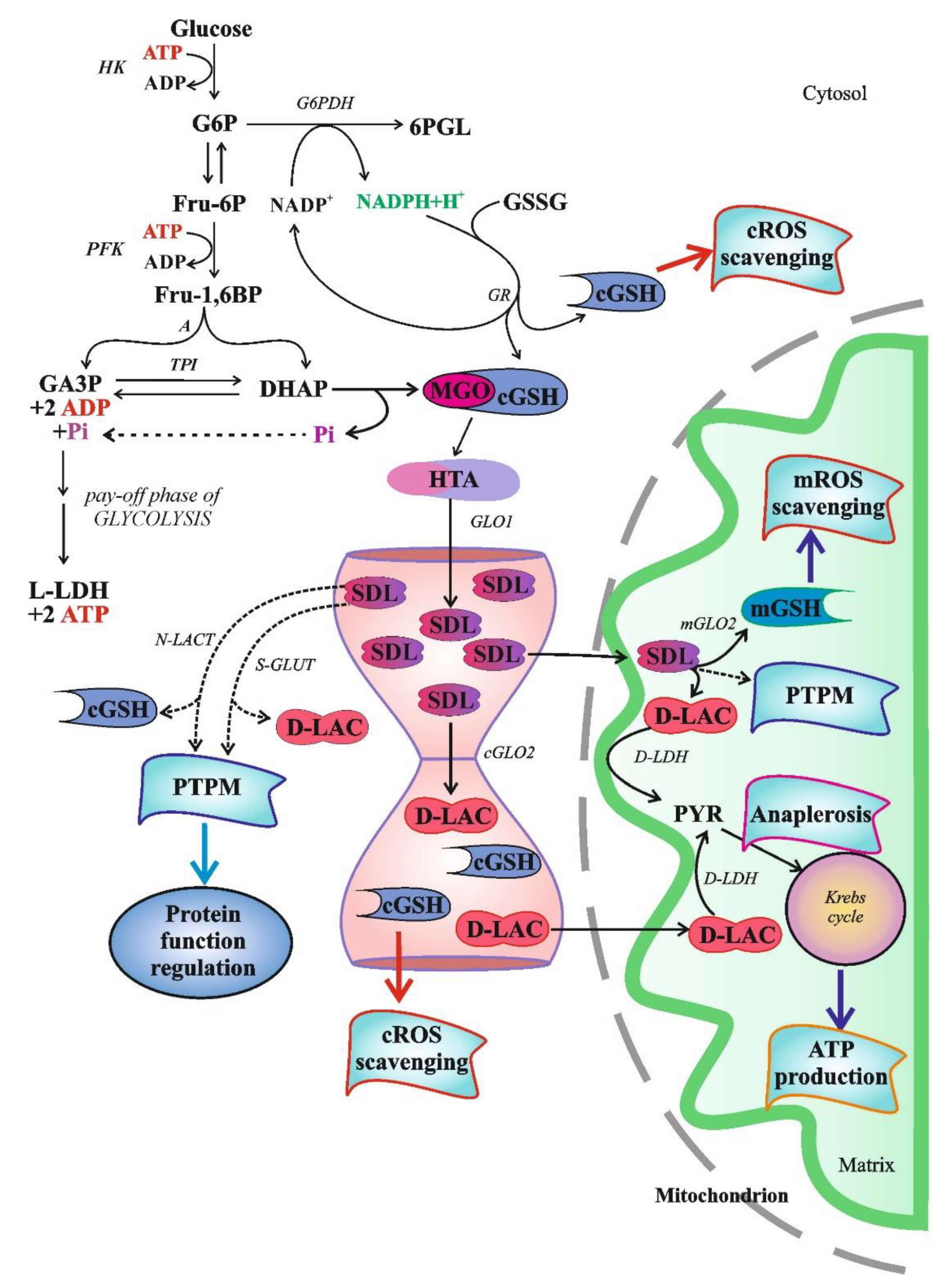
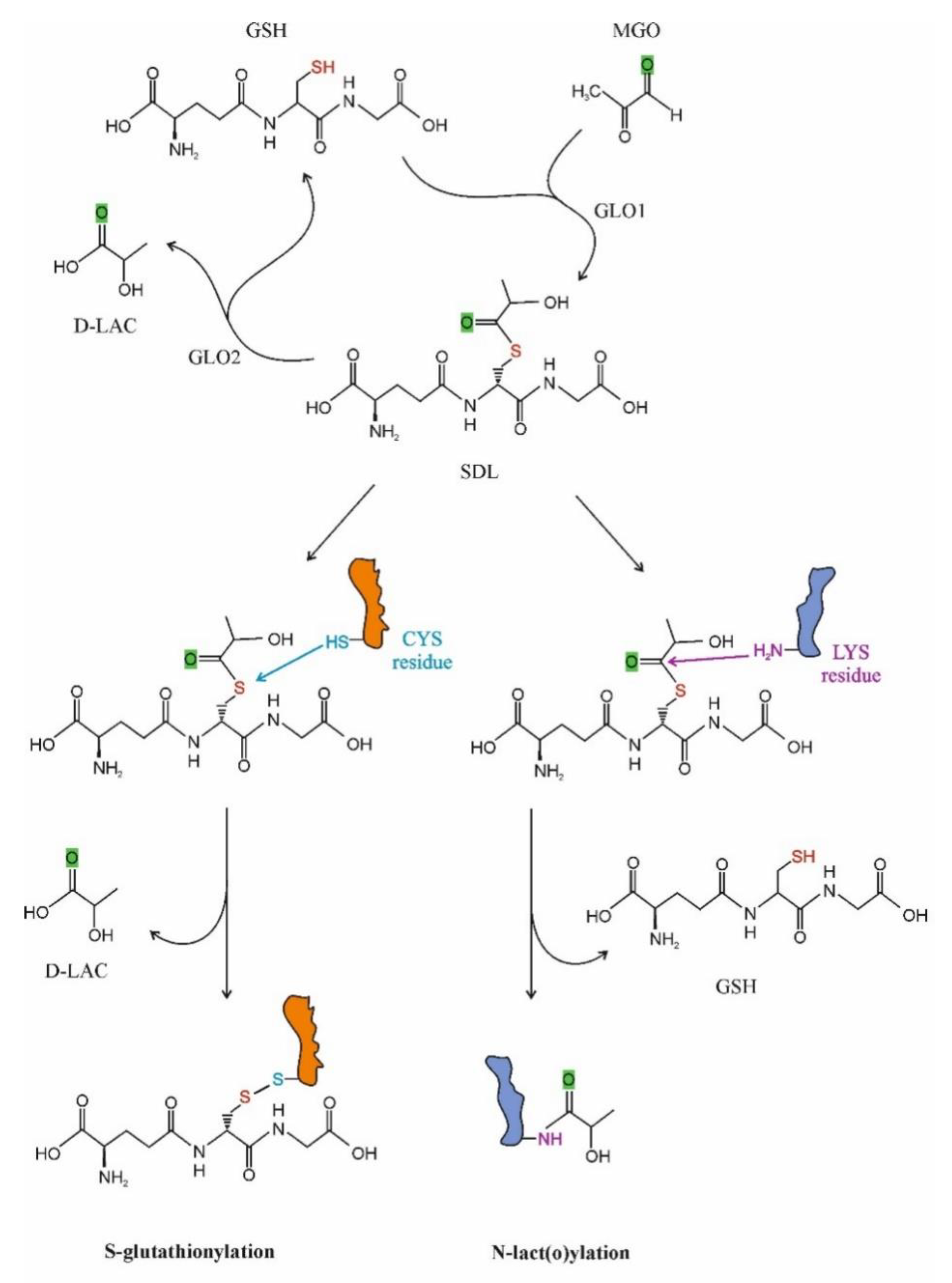
2.3.1. S-D-Lactoylglutathione Formation and Degradation
GLO1 (S-Lactoylglutathione Methlylglyoxal Lyase, EC 4.4.1.5)
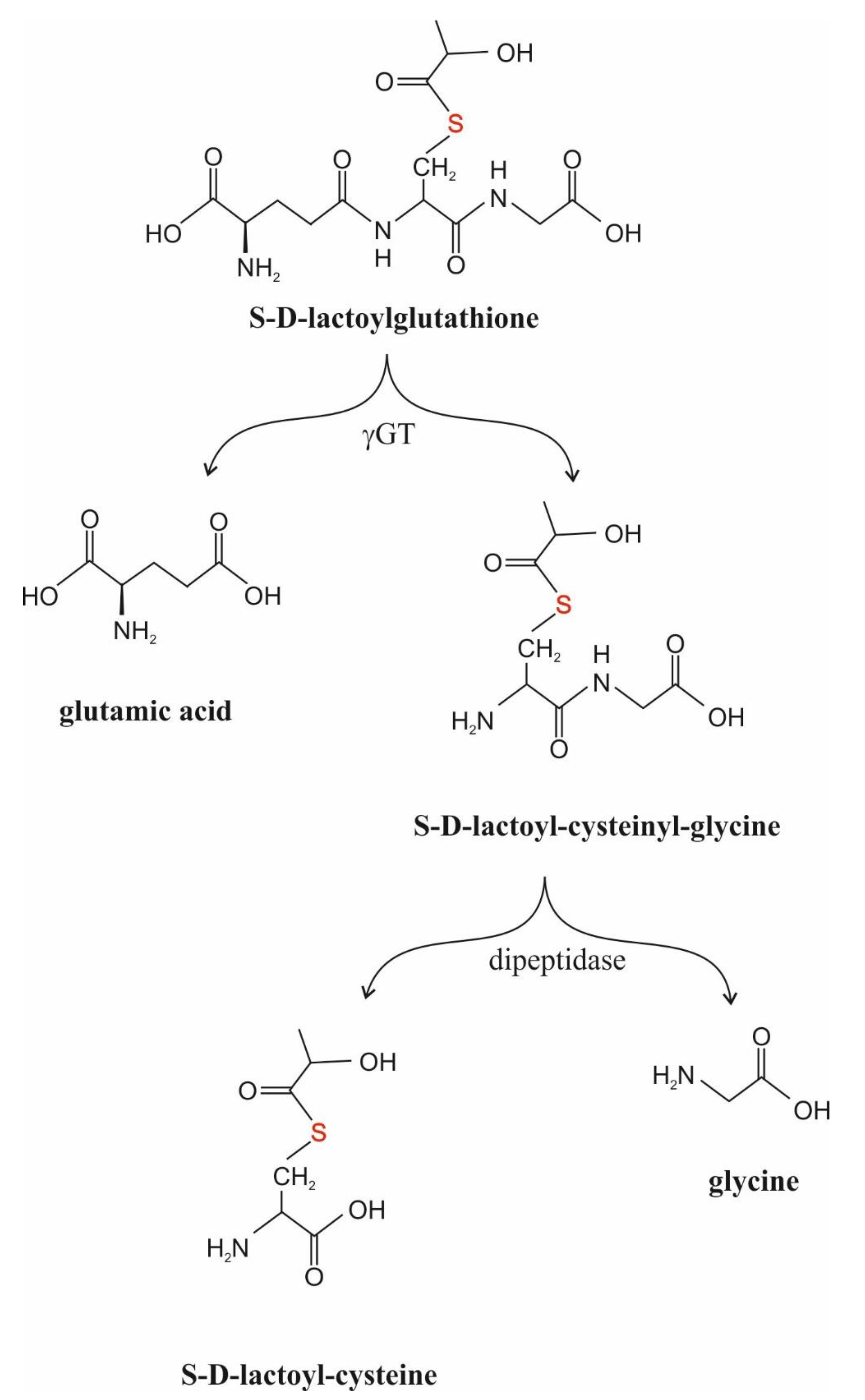
GLO2 (S-2-Hydroxyacylglutathione Hydrolase, EC 3.1.2.6)
sFGH (S-Formylglutathione Hydrolase, EC 3.1.2.12)
γGT (γ-Glutamyl-Transpeptidase, EC 2.3.2.2.)
Paroxysmal Non-Kinesigenic Dyskinesia (PNKD) Proteins
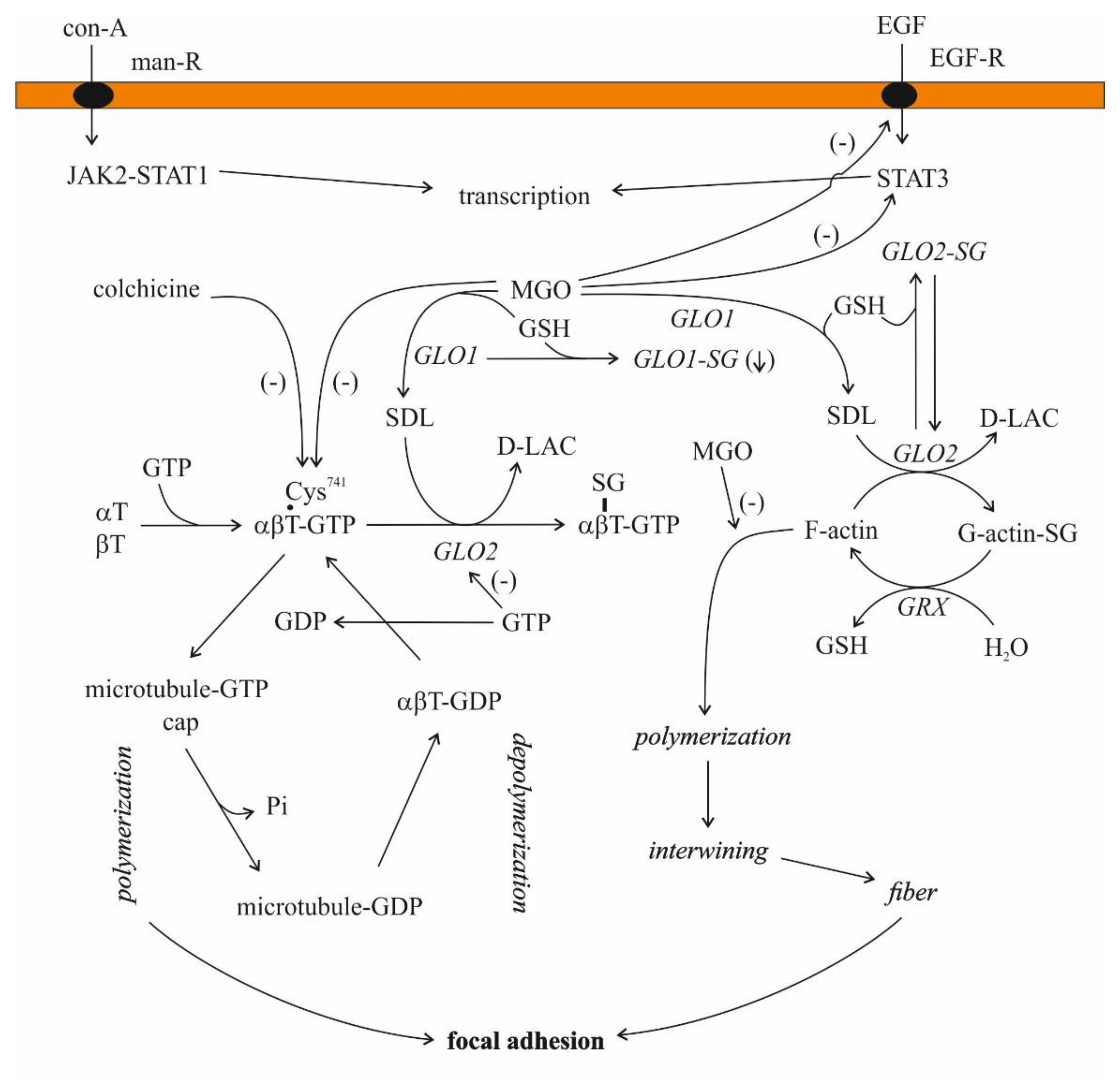
2.3.2. Regulation of Glyoxalases, with Glyoxalase II in the Focus
2.3.3. Thiols Replacing Glutathione in Glyoxalase Function
2.4. The Measurement of S-D-Lactoylglutathione in Biological Samples
3. Physiological Role of S-D-Lactoylglutathione
3.1. The Role of Intracellular S-D-Lactoylglutathione
3.1.1. Post-Translational Protein Modifications
3.1.2. Cytoskeleton Assembly
3.1.3. High-Energy Bond and S-D-Lactoylglutathione as an Energy Currency: Evolutionary Aspects
3.1.4. Glutathione and S-D-Lactoylglutathione Transport into Mitochondria
3.1.5. Role of S-D-Lactoylglutathione in Potassium Transport and Cellular Defense
4. The Role of Extracellular S-D-Lactoylglutathione
4.1. S-D-Lactoylglutathione, Cell Growth and Differentiation
4.2. S-D-Lactoylglutathione and Secretion
5. Brief Summary of the Possible Roles of S-D-Lactoylglutathione in Diseases
5.1. Hematological Disorders
5.2. Diabetes Mellitus
5.3. Thromboembolic Disorders
5.4. B1 Vitamin (Thiamine) Deficiency
5.5. Ketotic States Other Than Diabetes Mellitus
5.6. Paroxysmal Non-Kinesigenic Dyskinesia
5.7. Seroreactivity against Triose-Phosphate Isomerase
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Vander Jagt, D.L. The glyoxalase system. In Glutathione: Chemical, Biochemical and Medical Aspects; Part, A; Dolphin, D., Poulson, R., Avramovic, O., Eds.; John Wiley and Sons, Inc.: New York, NY, USA, 1989; pp. 597–641. [Google Scholar]
- Mannervik, B. Molecular enzymology of the glyoxalase system. Drug Metabol. Drug Interact. 2008, 23, 13–27. [Google Scholar] [CrossRef] [PubMed]
- De Bari, L.; Scirè, A.; Minnelli, C.; Cianfruglia, L.; Kalapos, M.P.; Armeni, T. Interplay among Oxidative Stress, Methylglyoxal Pathway and S-Glutathionylation. Antioxidants 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Creighton, D.J.; Migliorini, M.; Pourmotabbed, T.; Guha, M.K. Optimization of efficiency in the glyoxalase pathway. Biochemistry 1988, 27, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.E.; Thornalley, P.J. The hydrolysis of S-D-lactoylglutathione. Biochem. Soc. Transact. 1993, 21, 169S. [Google Scholar] [CrossRef]
- Yamazoye, S. Glyoxalase and its co-enzyme/III. The mechanism of the action of glutathione as the co-enzyme of glyoxalase. J. Biochem. 1936, 23, 319–334. [Google Scholar] [CrossRef]
- Racker, E. The mechanism of action of glyoxalase. J. Biol. Chem. 1951, 190, 685–696. [Google Scholar] [CrossRef]
- Uotila, L. Thioesters of glutathione. Meth. Enzymol. 1981, 77, 424–430. [Google Scholar] [CrossRef]
- Racker, E. Spectrophotometric measurements of the metabolic formation and degradation of thiol esters and enediol compounds. Biochim. Biophys. Acta 1952, 8, 577–578. [Google Scholar] [CrossRef]
- Piskorska, D.; Jerzykowsky, T.; Ostrowska, M. Synthesis of S-lactoyl-glutathione using glyoxalase I bound to Sepharose 4B. Experientia 1976, 32, 1382–1383. [Google Scholar] [CrossRef]
- Kosugi, N.; Inoue, Y.; Rhee, H.-I.; Murata, K.; Kimura, A. Production of S-lactoylglutathione by glycerol-adapted Saccharomyces cerevisiae and genetically engineered Escherichia coli cells. Appl. Microbiol. Biotechnol. 1988, 28, 263–267. [Google Scholar] [CrossRef]
- Clelland, J.D.; Thornalley, P.J. Synthesis of 14C-labelled methylglyoxal and S-D-lactoylglutathione. J. Label. Comp. Radiopharm. 1990, 28, 1455–1464. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993, 14, 287–371. [Google Scholar] [CrossRef]
- Kalapos, M.P. Methylglyoxal in living organisms/Chemistry, biochemistry, toxicology and clinical implications. Toxicol. Lett. 1999, 110, 145–175. [Google Scholar] [CrossRef]
- Inoue, Y.; Maeta, K.; Nomura, W. Glyoxalase system in yeasts: Structure, function, and physiology. Semin. Cell Dev. Biol. 2011, 22, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Rae, C.; Berners-Price, S.J.; Bulliman, B.T.; Kuchel, P.W. Kinetic analysis of the human erythrocyte glyoxalase system using 1H NMR and computer model. Eur. J. Biochem. 1990, 193, 83–90. [Google Scholar] [CrossRef]
- Mannervik, B.; Ridderström, M. Catalytic and molecular properties of glyoxalase I. Biochem. Soc. Transact. 1993, 21, 515–517. [Google Scholar] [CrossRef] [Green Version]
- Creighton, D.J.; Hamilton, D.S. Brief history of glyoxalase I and what we have learned about metal ion-dependent, enzyme-catalyzed isomerizations. Arch. Biochem. Biophys. 2001, 387, 1–10. [Google Scholar] [CrossRef]
- Honek, J.F. Glyoxalase biochemistry. BioMol Concepts 2015, 6, 401–414. [Google Scholar] [CrossRef]
- Kammerscheit, X.; Hecker, A.; Rouhier, N.; Chauvat, F.; Cassier-Chauvat, C. Methylglyoxal Detoxification Revisited: Role of Glutathione Transferase in Model Cyanobacterium Synechocystis sp. Strain PCC 6803. Mol. Biol. Physiol. 2020, 11, e00882-20. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef]
- Feierberg, I.; Cameron, A.D.; Åqvist, J. Energetics of the proposed rate-determining step of the glyoxalase I reaction. FEBS Lett. 1999, 453, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Lages, N.F.; Cordeiro, C.; Sousa Silva, M.; Ponces Freire, A.; Ferreira, A.E.N. Optimization of time-course experiments for kinetic model discrimination. PLoS ONE 2012, 7, e32749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellin, S.; Mannervik, B. Reversal of the reaction catalyzed by glyoxalase I/Calculation of the equilibrium constant for the enzymatic reaction. J. Biol. Chem. 1983, 258, 8872–8875. [Google Scholar] [CrossRef]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luengo, A.; Abbott, K.L.; Davidson, S.M.; Hosios, A.M.; Faubert, B.; Chen, S.H.; Freinkman, E.; Zachatias, L.G.; Mathews, T.P.; Clish, C.B.; et al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nat. Commun. 2019, 10, 5604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, S.S.; Doweyko, A.M.; Jordan, F. Glyoxalase I enzyme studies. 4 -General base catalyzed enediol proton transfer rearrangement of methyl- and phenylglyoxalglutathionylhemithiol acetal to L-lactoyl- and S-Mandeloylglutathione followed by hydrolysis. J. Am. Chem. Soc. 1978, 100, 5934–5939. [Google Scholar] [CrossRef]
- Vander Jagt, D.L. Glyoxalase II—Molecular characteristics, kinetics and mechanism. Biochem. Soc. Transact. 1993, 21, 522–526. [Google Scholar] [CrossRef]
- Rae, C.; Board, P.G.; Kuchel, P.W. Glyoxalase 2 deficiency in the erythrocytes of a horse: 1H NMR studies of enzyme kinetics and transport of S-lactoylglutathione. Arch. Biochem. Biophys. 1991, 291, 291–299. [Google Scholar] [CrossRef]
- Ball, J.C.; Vander Jagt, D.L. S-2-hydroxyacylglutathione hydrolase (glyoxalase II): Active-site mapping of a nonserine thiolesterase. Biochemistry 1981, 20, 899–905. [Google Scholar] [CrossRef]
- Antognelli, C.; Frosini, R.; Santolla, M.F.; Pierce, M.J.; Talesa, V.N. Oleuropein-induced apoptosis is mediated by mitochondrial glyoxalase 2 in NSCLC A549 cells: A mechanistic inside and possible novel nonenzymatic role for an ancient enzyme. Oxid Med. Cell Longev 2019, 2019, 8576961. [Google Scholar] [CrossRef] [Green Version]
- Principato, G.B.; Rosi, G.; Talesa, V.; Giovannini, E.; Uotila, L. Purification and characterization of two forms of glyoxalase II from the liver and brain of Wistar rats. Biochim. Biophys. Acta 1987, 911, 349–355. [Google Scholar] [CrossRef]
- Talesa, V.; Uotila, L.; Koivusalo, M.; Principato, G.; Giovannini, E.; Rosi, G. Demonstration of glyoxalase II in rat liver mitochondria. Partial purification and occurrence in multiple forms. Biochim. Biophys. Acta 1988, 955, 103–110. [Google Scholar] [CrossRef]
- Talesa, V.; Rosi, G.; Contenti, S.; Mangiabene, C.; Lupatelli, M.; Norton, S.J.; Giovannini, E.; Principato, G.B. Presence of glyoxalase II in mitochondria from Spinach leaves: Comparison with the enzyme from cytosol. Biochem. Int. 1990, 22, 1115–1120. [Google Scholar] [PubMed]
- Jassem, W.; Ciarimboli, C.; Cerioni, P.N.; Saba, V.; Norton, S.J.; Principato, G. Glyoxalase II and glutathione levels in rat liver mitochondria during cold storage in Euro-Collins and University of Wisconsin solutions. Transplantation 1996, 61, 1416–1420. [Google Scholar] [CrossRef]
- Bito, A.; Haider, M.; Handler, I.; Breitenbach, M. Purification and phenotypic analaysis of two glyoxalase II coding genes from Saccharomyces cerevisiae, GLO2 and GLO4, and intracellular localization of the corresponding proteins. J. Biol. Chem. 1997, 272, 21509–21519. [Google Scholar] [CrossRef] [Green Version]
- Maiti, M.K.; Krishnasamy, S.; Owen, H.A.; Makaroff, C.A. Molecular characterization of glyoxalase II from Arabidopsis thaliana. Plant Mol. Biol. 1997, 35, 471–481. [Google Scholar] [CrossRef]
- Bito, A.; Haider, M.; Briza, P.; Strasser, P.; Breitenbach, M. Heterologous expression, purification, and kinetic comparison of the cytoplasmic and mitochondrial glyoxalase II enzymes, Glo2p and Glo4p, from Saccharomyces cerevisiae. Protein Expr. 1999, 17, 456–464. [Google Scholar] [CrossRef]
- Cordell, P.A.; Futers, T.S.; Grant, P.J.; Pease, R.J. The human hydroxyglutathione hydrolase (HAGH) gene encodes both cytosolic and mitochondrial forms of glyoxalase II. J. Biol. Chem. 2004, 279, 28653–28661. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Bisht, R.; Roy, S.D.; Sopory, S.K.; Bhalla-Sarin, N. Cloning and characterization of a mitochondrial glyoxalase II from Brassica juncea that is upregulated by NaCl, Zn, and ABA. Biochem. Biophys. Res. Commun. 2005, 336, 813–819. [Google Scholar] [CrossRef]
- Xue, M.-Z.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Urscher, M.; Alisch, R.; Deponte, M. The glyoxalase system of malaria parasites—Implications for cell biology and general glyoxalase research. Semin. Cell Dev. Biol. 2011, 22, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Ferri, I.; Bellezza, G.; Siccu, P.; Love, H.D.; Talesa, V.N.; Sidoni, A. Glyoxalase 2 drives tumorigenesis in human prostate cells in a mechanism involving androgen receptor and p53-p21 axis. Mol. Carcinog. 2017, 56, 2112–2128. [Google Scholar] [CrossRef] [PubMed]
- Uotila, L.; Koivusalo, M. Purification and properties of S-formylglutathione hydrolase from human liver. J. Biol. Chem. 1974, 248, 7664–7673. [Google Scholar] [CrossRef]
- Gonzalez, C.F.; Proudfoot, M.; Brown, G.; Korniyenko, Y.; Mori, H.; Savchenko, A.V.; Yakunin, A.F. Molecular basis of formaldehyde detoxification/Characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J. Biol. Chem. 2006, 281, 14514–14522. [Google Scholar] [CrossRef] [Green Version]
- Van Straaten, K.E.; Gonzalez, C.F.; Valladares, R.B.; Xu, X.; Savchenko, A.V.; Sanders, D.A.R. The structure of S-formylglutathione hydrolase from Agrobacterium tumefaciens. Protein Sci. 2009, 18, 2196–2202. [Google Scholar] [CrossRef] [Green Version]
- Apeshiotis, F.; Bender, K. Evidence that S-formylglutathione hydrolase and esterase-D polymorphisms are identical. Hum. Genet. 1986, 74, 176–177. [Google Scholar] [CrossRef]
- Eiberg, H.; Mohr, J. Identity of the polymorphisms for esterase-D and S-formylglutathione hydrolase in red-blood-cells. Hum. Genet. 1986, 74, 175–176. [Google Scholar] [CrossRef]
- Tate, S.S. Interaction of γ-glutamyl transpeptidase with S-acyl derivatives of glutathione. FEBS Lett. 1975, 54, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Meister, A.; Tate, S.S.; Griffith, O.W. γ-glutamyltransferase. Meth. Enzymol. 1981, 77, 237–253. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glutathione-dependent detoxification of α-oxoaldehydes by glyoxalase system: Involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem. Biol. Interact. 1998, 111–112, 137–151. [Google Scholar] [CrossRef]
- Whitfield, J.B. Gamma Glutamyl Transferase. Crit. Rev. Clin. Lab. Sci. 2001, 38, 263–355. [Google Scholar] [CrossRef] [PubMed]
- Pompella, A.; De Tata, V.; Paolicchi, A.; Zunino, F. Expression of γ-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem. Pharmacol. 2005, 71, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.M.; Cordeiro, C.; Freire, A.P. Glyoxalase II in Saccharomyces cerevisiae: In situ kinetics using the 5,5′-dithiobis(2-nitrobenzoic acid) assay. Arch. Biochem. Biophys. 1999, 366, 15–20. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Xu, Y.; Huang, Y.; Ahn, A.H.; Auburger, G.W.J.; Pandolfo, M.; Kwiecinski, H.; Grimes, D.A.; Lang, A.E.; Nielsen, J.E.; et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum. Mol. Genet. 2004, 13, 3161–3170. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Lee, H.-Y.; Rawson, J.; Ojha, S.; Babbitt, P.; Fu, Y.-H.; Ptáĉek, L.J. Mutations in PNKD causing paroxysmal dyskinesia alters protein cleavage and stability. Hum. Mol. Genet. 2011, 20, 2322–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettinati, I.; Brem, J.; Lee, S.Y.; McHugh, P.J.; Schofield, C.J. The chemical biology of human metallo-β-lactamase fold proteins. Trends Biochem. 2016, 41, 338–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erro, R.; Bhatia, K.P.; Espay, A.J.; Striano, P. The epileptic and non-epileptic spectrum of paroxysmal dyskinesias: Channelopathies, synaptopathies, and transportopathies. Mov. Disord. 2017, 32, 310–318. [Google Scholar] [CrossRef]
- Ghezzi, D.; Viscomi, C.; Ferlini, A.; Gialandi, F.; Mereghetti, P.; DeGrandis, D.; Zeviani, M. Paroxysmal non-kinesigenic dyskinesia is caused by mutations of the MR-1 mitochondrial targeting sequence. Hum. Mol. Genet. 2009, 18, 1058–1064. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; He, H.; Liu, H.; Zhang, C.; Zhao, W.; Shao, R. Phosphorylation of myofibrillogenesis regulator-1 activates the MAPK signaling pathway and induces proliferation and migration in human breast cancer MCF7 cells. FEBS Lett. 2014, 588, 2903–2910. [Google Scholar] [CrossRef] [Green Version]
- Birkenmeier, G.; Stegemann, C.; Hoffmann, R.; Günther, R.; Huse, K.; Birkemeyer, C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS ONE 2010, 5, e10399. [Google Scholar] [CrossRef] [Green Version]
- Antognelli, C.; Talesa, V.N. Glyoxalases in urological malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla-Pareek, S.L.; Kaur, C.; Kumar, B.; Pareek, A.; Sopory, S.K. Reassessing plant glyoxalases: Large family and expanding functions. New Phytol. 2020, 227, 714–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenstern, J.; Campos, M.; Nawroth, P.; Fleming, T. The glyoxalase system—New insights into an ancient metabolism. Antioxidants 2020, 9, 939. [Google Scholar] [CrossRef] [PubMed]
- Dafre, A.L.; Goldberg, J.; Wang, T.; Spiegel, D.A.; Maher, P. Methylglyoxal, the foe and friend of glyoxalase and Trx/TrxR systems in HT22 nerve cells. Free Radic. Biol. Med. 2015, 89, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Chen, X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J. Biol. Chem. 2006, 281, 26702–26713. [Google Scholar] [CrossRef] [Green Version]
- Anaki, N.; Morimasa, T.; Salai, T.; Tokuoh, H.; Yunoue, S.; Kamo, M.; Miyazaki, K.; Abe, K.; Saya, H.; Tsugita, A. Comparative analysis of brain proteins from p53-deficient mice by two-dimensional electrophoresis. Electrophoresis 2000, 21, 1880–1889. [Google Scholar] [CrossRef]
- Gillespie, E. The tumor promoting phorbol diester, 12-O-tetradecanoylphorbol-13-acetate (TPA) increases glyoxalase I and decreases glyoxalase II in human polymorphonuclear leukocytes. Biochem. Biophys. Res. Commun. 1981, 98, 463–470. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Bellavite, P. Modification of the glyoxalase system during functional activation of human neutrophils. Biochim. Biophys. Acta 1987, 931, 120–129. [Google Scholar] [CrossRef]
- Murata, K.; Sato, N.; Inoue, Y.; Kimura, A. S-D-lactoylglutathione: Control of the cellular level by a yeast glyoxalase system. Agric. Biol. Chem. 1989, 53, 1999–2000. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Inoue, Y.; Rhee, H.; Kimura, A. 2-oxoaldehyde metabolism in microorganisms. Can. J. Microbiol. 1989, 35, 423–431. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Della Bianca, V.; Bellavite, P.; Rossi, F. S-D-lactoylglutathione in resting and activated human neutrophils. Biochem. Biophys. Res. Commun. 1987, 145, 769–774. [Google Scholar] [CrossRef]
- Inoue, Y.; Choi, B.-Y.; Murata, K.; Kimura, A. Sexual response in Saccharomyces cerevisiae: Alteration of enzyme activity in the glyoxalase system by mating factor. Biochem. Biophys. Res. Commun. 1989, 165, 1091–1095. [Google Scholar] [CrossRef]
- Inoue, Y.; Choi, B.-Y.; Murata, K.; Kimura, A. Sexual response of Saccharomyces cerevisiae: Phosphorylation of yeast glyoxalase I by a cell extract of mating factor-treated cells. J. Biochem. 1990, 108, 4–6. [Google Scholar] [CrossRef]
- Kimura, A.; Inoue, Y. Glyoxalase I in microorganisms: Molecular characteristics, genetics and biochemical regulation. Biochem. Soc. Transact. 1993, 21, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Herreweghe, F.; Mao, J.; Chaplen, F.W.R.; Grooten, J.; Gevaert, K.; Vandekerckhove, J.; Vancompernolle, K. Tumor Necrosis Factor-Induced (cytokine) Modulation of Glyoxalase I Activities Through Phosphorylation by PKA Results in Cell Death and Is Accompanied by the Formation of a Specific Methylglyoxal-Derived AGE. Proc. Natl. Acad. Sci. USA 2002, 99, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Hemptine, V.; Rondas, D.; Toepoel, M.; Vancompernolle, K. Tumour necrosis factor induces phosphorylation primarily of the nitric-oxide-responsive form of glyoxalase I. Biochem. J. 2007, 487, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Hemptine, V.; Rondas, D.; Toepoel, M.; Vancompernolle, K. Phosphorylation on Thr-106 and NO-modification of glyoxalase I suppress the TNF-induced transcriptional activity of NF-κB. Mol. Cell Biochem. 2009, 325, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Teijero, J.M.; Marini, P.E. Hormone-regulated PKA activity in porcine oviductal epithelial cells. Cell Tissue Res. 2020, 380, 357–367. [Google Scholar] [CrossRef]
- Mitsumoto, A.; Kim, K.-R.; Oshima, G.; Kunimoto, M.; Okawa, K.; Iwamatsu, A.; Nakagawa, Y. Glyoxalase I is a novel nitric-oxide-responsive protein. Biochem. J. 1999, 344, 837–844. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Alam, M.M.; Fujita, M. Exogenous nitric oxide alleviates high temperature induced oxidative stress in wheat (Triticum aestivum L.) seedlings by modulating the antioxidant defense and glyoxalase system. Aust. J. Crop. Sci. 2012, 6, 1314–1323. [Google Scholar]
- Hasanuzzaman, M.; Nahar, K.; Hossain, M.S.; Islam, A.; Parvin, K.; Fujita, M. Nitric oxide pretreatment enhances antioxidant defense and glyoxalase systems to confer PEG-induced oxidative stress in rapeseed. J. Plant Interact. 2017, 12, 323–331. [Google Scholar] [CrossRef]
- Kehr, S.; Jortzik, E.; Delahunty, C.; Yates, J.R.; Rahifs, S.; Becker, K. Protein S-glutathionylation in malaria parasites. Antioxid. Redox Signal. 2011, 15, 2855–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchers, J.; Dirdjaja, N.; Ruppert, T.; Krauth-Siegel, R.L. Glutathionylation of trypanosomal thiol redox proteins. J. Biol. Chem. 2007, 282, 8678–8694. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Liebau, E.; Walter, R.D.; Krauth-Siegel, E.L. Thiol-based redox metabolism of protozoan parasites. Trends Parasitol. 2003, 19, 320–328. [Google Scholar] [CrossRef]
- Wyllie, S.; Fairlamb, A.H. Methylglyoxal metabolism… in trypanosomes and leishmania. Semin. Cell Dev. Biol. 2011, 22, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brophy, P.M.; Crowley, P.; Barrett, J. Relative distribution of glutathione transferase, glyoxalase I and glyoxalase II in helminthes. Int. J. Parasitol. 1990, 20, 259–261. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Patterson, S.; Fairlamb, A.H. A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J. 2009, 276, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Arbach, M.; Roberts, A.; Macdonald, C.J.; Groom, M.; Hamilton, C.J. Biophysical features of bacillithiol, the glutathione surrogate of Bacillus subtilis and other Firmucites. Chembiochem 2013, 14, 2160–2168. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, G.P.; McLaggan, D.; Booth, I.R. Potassium channel activation by glutathione-S-conjugates in Escherichia coli: Protection against methylglyoxal is mediated by cytoplasmic acidification. Mol. Microbiol 1995, 17, 1025–1033. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Dusi, R.; Hamilton, C.J.; Helmann, J.D. Methylglyoxal resistance in Bacillus subtilis: Contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014, 91, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Loi, V.V.; Rossius, M.; Antelmann, H. Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suttisansanee, U.; Honek, J.F. Preliminary characterization of a Ni2+-activated and mycothiol-dependent glyoxalase I enzyme from Streptomyces coelicolor. Inorganics 2019, 7, 99. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its analogs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef] [Green Version]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Kimura, M.; Queliconi, B.B.; Kojima, W.; Mishima, M.; Takagi, K.; Koyano, F.; Yamono, K.; Mizushima, T.; Ito, Y.; et al. Parkinson’s disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci. Rep. 2017, 7, 12816. [Google Scholar] [CrossRef] [Green Version]
- Klotzsch, H.; Bergmeyer, H.-U. Methylglyoxal. and Glutathione. In Methods in Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic Press Inc.: New York, NY, USA, 1963; pp. 283–284, 363–366. [Google Scholar]
- Warholm, M.; Guthenberg, C.; von Bahr, C.; Mannervik, B. Glutathione transferases from human liver. Meth. Enzymol 1985, 113, 499–503. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Hooper, N.I.; Jennings, P.E.; Florkowski, C.M.; Jones, A.F.; Lunec, J.; Barnett, A.H. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res. Clin. Pract. 1989, 7, 115–120. [Google Scholar] [CrossRef]
- Hooper, N.I.; Tisdale, M.J.; Thornalley, P.J. Modification of the glyoxalase system in human HL60 promyelocytic leukaemia cell during differentiation to neutrophils in vitro. Biochim. Biophys. Acta 1988, 966, 362–369. [Google Scholar] [CrossRef]
- Hooper, N.I.; Tisdale, M.J.; Thornalley, P.J. Glyoxalase activity and cell proliferation in Burkitt’s lymphoma and transformed lymphoblast cells in vitro. Cell Mol. Biol. 1988, 34, 399–405. [Google Scholar]
- Thornalley, P.J.; Tisdale, M.J. Inhibition of proliferation of human promyelocytic leukaemia HL60 cells by S-D-lactoylglutathione in vitro. Leuk. Res. 1988, 12, 897–904. [Google Scholar] [CrossRef]
- Kalapos, M.P.; Garzó, T.; Antoni, F.; Mandl, J. Accumulation of S-D-lactoylglutathione and transient decrease of glutathione level caused by methylglyoxal load in isolated hepatocytes. Biochim. Biophys. Acta 1992, 1135, 159–164. [Google Scholar] [CrossRef]
- McLellan, A.C.; Phillips, S.A.; Thornalley, P.J. The assay of S-D-lactoylglutathione in biological systems. Anal. Biochem. 1993, 211, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, G.; Buzzi, E.; Aprile, B. S-D-lactoylglutathione accumulation in activated human platelets. Int. J. Biochem. 1993, 25, 1565–1570. [Google Scholar] [CrossRef]
- Uchino, E.; Fukushima, T.; Tsunoda, M.; Santa, T.; Imai, K. Determination of rat blood S-D-lactoylglutathione by a column-switching high-performance liquid chromatography with a precolumn fluorescence derivatization with 4-fluoro-7-nitro-2,1,3-benzoxadiazole. Anal. Biochem. 2004, 330, 186–192. [Google Scholar] [CrossRef]
- Edwards, L.; Clelland, J.D.; Thornalley, P.J. Characteristics of the inhibition of human promyelocytic leukaemia HL60 cell growth by S-D-lactoylgluthathione in vitro. Leuk. Res. 1993, 17, 305–310. [Google Scholar] [CrossRef]
- Leoncini, G.; Buzzi, E. Thrombin induces S-D-lactoylglutathione accumulation by enhancing platelet glycolytic pathway. Int. J. Biochem. 1994, 26, 661–665. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 1994, 87, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-X.; Zheng, H.; Yao, X.; Liu, X.-W.; Zhu, H.-J.; Yin, C.-L.; Liu, X.; Mo, Y.-Y.; Huang, H.-M.; Cheng, B.; et al. Comparative analysis of the compatibility effects of Danggui-Sini Decoction on a blood stasis syndrome rat model using untargeted metabolomics. J. Chromatogr. 2019, 1105, 164–175. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Activity, regulation, copy number and function int he glyoxalase system. Biochem. Soc. Transact. 2014, 42, 419–424. [Google Scholar] [CrossRef]
- Shin, M.J.; Edinger, J.W.; Creighton, D.J. Diffusion-dependent kinetic properties of glyoxalase I and estimates of the steady-state concentrations of glyoxalase pathway intermediates in glycolyzing erythrocytes. Eur. J. Biochem. 1997, 244, 852–857. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Transact. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L.; Hassenbrook, R.K.; Hunsaker, L.A.; Brown, W.M.; Royer, R.E. Metabolism of the 2-oxoaldehyde methylglyoxal by aldose reductase and by glyoxalase-I: Roles for glutathione in both enzymes and implications for diabetic complications. Chem. Biol. Interact. 2001, 130–132, 549–562. [Google Scholar] [CrossRef]
- Kalapos, M.P.; Wu, L. Can methylglyoxal be oxidized to CO2 in vascular smooth muscle cells? J. Investig. Biochem. 2014, 3, 149–153. [Google Scholar] [CrossRef]
- Knorre, D.G.; Kudryashova, N.V.; Godovokova, T.S. Chemical and functional aspects of posttranslational modification of proteins. Acta Nat. 2009, 3, 29–51. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, V.P.A.; Hool, L.C. Glutathionylation of the L-type Ca2+ channel in oxidative stress-induced pathology in the hearth. Int. J. Mol. Sci. 2014, 13, 19203–19225. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jin, X.; Jiang, C. S-glutathionylation of ion channels: Insights into the regulation of channel functions, thiol modification crosstalk, and mechanosensing. Antioxid. Redox Signal. 2014, 20, 6. [Google Scholar] [CrossRef]
- Wilson, C.; González-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Sciré, A.; Tanfani, F.; Saccucci, F.; Bertoli, E.; Principato, C. Specific interaction of cytosolic and mitochondrial glyoxalase II with acidic phospholipids in form of liposomes results in the inhibition of the cytosolic enzyme only. Proteins 2000, 41, 33–39. [Google Scholar] [CrossRef]
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianfruglia, L.; Galeazzi, R.; Massaccesi, L.; Spaccini, R.; Caniglia, M.L.; Principato, G.; Armeni, T. Glyoxalase II promotes “in vitro” S-glutathionylation. Free Radic. Biol. Med. 2014, 75, S26–S27. [Google Scholar]
- Dominko, K.; Ðikic, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arch. Hig. Rada Toksikol. 2018, 69, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Ercolani, L.; Sciré, A.; Galeazzi, R.; Massaccesi, L.; Cianfruglia, L.; Amici, A.; Piva, F.; Urbanelli, L.; Emilliani, C.; Principato, G.; et al. A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochem. Funct. 2016, 34, 620–627. [Google Scholar] [CrossRef]
- Galeazzi, R.; Laudadio, E.; Falconi, E.; Massaccesi, L.; Ercolani, L.; Mobbili, G.; Minnelli, C.; Sciré, A.; Cianfruglia, L.; Armeni, T. Protein-protein interactions of human glyoxalase II: Findings of a reliable docking protocol. Org. Biomol. Chem. 2018, 16, 5167–5177. [Google Scholar] [CrossRef]
- Darnell, J.; Lodish, H.; Baltimore, D. Molecular Cell Biology; Scientific American Books, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Tang, D.D.; Gerlach, B.D. The roles and regulation of actin cytoskeleton, intermediate filaments and microtubules in smooth muscle cell migration. Respir. Res. 2017, 18, 54. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [Green Version]
- Sakai, J.; Li, J.; Subramanian, K.K.; Mondal, S.; Bajrami, B.; Hattori, H.; Jia, Y.; Dickinson, B.C.; Zhong, J.; Ye, K.; et al. Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity 2012, 37, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Hoogewijs, K.; Logan, A.; Hall, A.R.; Ding, S.; Fearnley, I.M.; Murphy, M.P. Non-enzymatic N-acetylation of lysine residues by acetyl-CoA often occurs via a proximal S-acetylated thiol intermediate sensitive to glyoxalase II. Cell Rep. 2017, 18, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.-N.; Luo, Y.; Yang, Y.-H.; Fu, J.-T.; Geng, X.-M.; Shi, J.-P.; Yang, J. Lactylation, a Novel Metabolic Reprogramming Code: Current Status and Prospects. Front. Immunol. 2021, 12, 688910. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, D.O.; Jennings, E.O.; Anderson, C.C.; Marentette, J.O.; Shi, T.; Schou Oxvig, A.-M.; Streeter, M.D.; Johannsen, M.; Spiegel, D.A.; Chapman, E.; et al. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem. Biol. 2020, 27, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Khadka, S.; Barekatain, Y.; Muller, F.L. Re-Evaluating the Mechanism of Histone Lactylation. 2020. Available online: https://www.researchgate.net/publication/341582046 (accessed on 1 May 2020). [CrossRef]
- Kulkarni, C.A.; Brookes, P.S. Many Routes from Glycolysis to Histone PTMs. 2020. Nature “Matters Arising” response to: Zhang et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Conde, C.; Cóceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Macconi, R.B.; Vera, J.C.; Slebe, J.C. Arginyl residues involvement in the microtubule assambly. Arch. Biochem. Biophys. 1981, 297, 248–255. [Google Scholar] [CrossRef]
- Kalapos, M.P. Methylglyoxal toxicity in mammals. Toxicol. Lett. 1994, 73, 3–24. [Google Scholar] [CrossRef]
- Miglietta, A.; Gabriel, L. Methylglyoxal-tubulin interaction: Studies on the aldehyde effects on hepatoma, liver and purified microtubular protein. Res. Commun. Chem. Pathol. Pharmacol. 1986, 51, 245–260. [Google Scholar]
- Fésűs, L.; Muszbek, L.; Laki, K. The effect of methylglyoxal on actin. Biochem. Biophys. Res. Commun. 1981, 99, 617–622. [Google Scholar] [CrossRef]
- Dianzani, M.U. Biological activity of methylglyoxal and related aldehydes. In Submolecular Biology and Cancer; CIBA Foundation Series 67; Elsevier/North Holland: New York, NY, USA, 1979; pp. 245–270. [Google Scholar]
- Gillespie, E. Cell-free microtubule assembly: Evidence for control by glyoxalase. Fed. Proc. 1975, 34, 541. [Google Scholar]
- Clelland, J.D.; Thornalley, P.J. The potentiation of GTP-dependent assembly of microtubules by S-D-lactoylglutathione. Biochem. Soc. Transact. 1993, 21, 160S. [Google Scholar] [CrossRef] [PubMed]
- Di Simplicio, P.; Vignani, R.; Talesa, V.; Principato, G. Evidence of glyoxalase II activity associated with microtubule polymerization in bovine brain. Pharmacol. Res. 1990, 22, 172. [Google Scholar] [CrossRef]
- Norton, S.J.; Elia, A.C.; Chyan, M.K.; Gillis, G.; Frenzel, C.; Principato, G.B. Inhibitors and inhibition studies on mammalian glyoxalase II activity. Biochem. Soc. Transact. 1993, 21, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Seefeldt, T.; Young, A.; Zhan, X.; Zhao, Y.; Ruffolo, J.; Kaushik, R.S.; Guan, X. Microtubule S-glutathionylation as a potential approach for antimitotic agents. BMC Cancer 2012, 12, 245. [Google Scholar] [CrossRef] [Green Version]
- Kalapos, M.P. Possible evolutionary role of methylglyoxalase pathway/Anaplerotic route for surface metabolists. J. Theor. Biol. 1997, 188, 201–206. [Google Scholar] [CrossRef]
- Kalapos, M.P. From mineral support to enzymatic catalysis/Further assumptions for the evolutionary history of glyoxalase system. J. Theor. Biol. 1998, 193, 91–98. [Google Scholar] [CrossRef]
- Kalapos, M.P. A theoretical approach to the link between oxido-reductions and pyrite formation in the early stage of evolution. Biochim. Biophys. Acta 2002, 1553, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Racker, E. Mechanisms in Bioenergetics; Academic Press Inc.: New York, NY, USA, 1965. [Google Scholar]
- Armeni, T.; Cianfruglia, L.; Piva, F.; Urbanelli, L.; Cinaglia, M.L.; Pugnaloni, A.; Principato, G. S-D-lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014, 67, 451–459. [Google Scholar] [CrossRef]
- De Bari, L.; Atlante, A.; Armeni, T.; Kalapos, M.P. Synthesis and metabolism of methylglyoxal, S-D-lactoylglutathione and D-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Ageing Res. Rev. 2019, 53, 100915. [Google Scholar] [CrossRef] [PubMed]
- Wendler, A.; Irsch, T.; Rabbani, N.; Thornalley, P.J.; Krauth-Siegel, R.L. Glyoxalase II does not support methylglyoxal detoxification but serves as a general trypanothione thioesterase in African Trypanosomes. Mol. Biochem. Parasitol. 2009, 163, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid. Med. Cell Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef] [PubMed]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its s.selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Borysiuk, K.; Ostaszewska-Gugajska, M.; Vaultier, M.-N.; Hasenfratz-Sauder, M.-P.; Szal, B. Enhanced formation of methylglyoxla-derived advanced glycation end products in Arabidopsis under ammonium nutrition. Front. Plant Sci. 2018, 9, 667. [Google Scholar] [CrossRef]
- Chaplen, F.W.R.; Fahl, W.E.; Cameron, D.C. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal. Biochem. 1996, 238, 171–178. [Google Scholar] [CrossRef]
- Ferguson, G.P. Protective mechanisms against toxic electrophiles in Escherichia coli. Trends Microbiol. 1999, 7, 242–247. [Google Scholar] [CrossRef]
- Ferguson, G.P.; Booth, I.R. Importance of glutathione for growth and survival of Escherichia coli cells: Detoxification of methylglyoxal and maintenance of intracellular K+. J. Bact. 1998, 180, 4314–4318. [Google Scholar] [CrossRef] [Green Version]
- Booth, I.R.; Ferguson, G.P.; Miller, S.; Li, C.; Gunasekera, B.; Kinghorn, S. Bacterial production of methylglyoxal: A survival strategy or death by misadventure? Biochem. Soc. Transact. 2003, 31, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, G.P.; Munor, A.W.; Douglas, R.M.; McLaggan, D.; Booth, I.R. Activation of potassium channels during metabolite detoxification in Escherichia coli. Mol. Microbiol. 1993, 9, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.J.; Ness, L.S.; Ferguson, G.P.; Booth, I.R. The role of glyoxalase I in the detoxification of methylglyoxal and in the activation of the KefB K+ efflux system in Escherichia coli. Mol. Microbiol. 1998, 27, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Chaudhuri, D.; Balakrishnan, A.; Chakrawortty, D. Salmonella methylglyoxal detoxification by STM3117-encoded lactoylglutathione lyase affects virulence in coordination with Salmonella pathogenicity island 2 and phagosomal acidification. Microbiology 2014, 160, 1999–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozyamak, E.; Black, S.S.; Walker, C.A.; MacLean, M.J.; Bartlett, W.; Miller, S.; Booth, I.R. The critical role of S-D-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol. Microbiol. 2010, 78, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, L.; Booth, I.R. Different foci for the regulation of the activity of the KefB and KefC glutathione-gated K+ efflux systems. J. Biol. Chem. 1999, 274, 9524–9530. [Google Scholar] [CrossRef] [Green Version]
- McKie, J.H.; Douglas, K.T. Structural relationships between glyoxalase I and membrane transport proteins. Biochem. Soc. Transact. 1993, 21, 540–544. [Google Scholar] [CrossRef] [Green Version]
- Funderburk, L.H.; Aldwin, L.; Jencks, W.P. Mechanisms of general acid and base catalysis of the reactions of water and alcohols with formaldehyde. J. Am. Chem. Soc. 1978, 100, 5444–5459. [Google Scholar] [CrossRef]
- Morrissey, A.; Rosner, E.; Lanning, J.; Parachuru, L.; Chowdhury, P.D.; Han, S.; Lopez, G.; Tong, X.Y.; Yoshida, H.; Nakamura, T.Y.; et al. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2015, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Kalapos, M.P. Possible mechanism for the effect of ketogenic diet in cases of uncontrolled seizures / Reconsideration of acetone theory. Med. Hypotheses 2007, 68, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, W.; Cui, N.; Wu, Z.; Jiang, C. Oxidative stress inhibits vascular KATP channels by S-Glutathionylation. J. Biol. Chem. 2010, 285, 38641–38648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shi, W.; Chen, X.; Cui, N.; Konduru, A.S.; Shi, Y.; Trower, T.; Zhang, S.; Jiang, C. Molecular basis and structural insight of vascular KATP channel gating by S-Glutathionylation. J. Biol. Chem. 2011, 286, 9298–9307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, S.; Konduru, A.S.; Zhang, S.; Trower, T.; Shi, W.; Cui, N.; Yu, L.; Wang, Y.; Zhu, D.; et al. Prolonged exposure to methylglyoxal causes disruption of vascular KATP channel by mRNA instability. Am. J. Physiol. Cell Physiol. 2012, 303, C1045–C1054. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Konduru, A.S.; Cui, N.; Trower, T.C.; Shi, W.; Shi, Y.; Jiang, C. Acute exposure of methylglyoxal leads to activation of KATP channels expressed in HEK293 cells. Acta Pharmacol. Sin. 2014, 35, 58–64. [Google Scholar] [CrossRef]
- Mukohda, M.; Yamawaki, H.; Nomura, H.; Okada, M.; Hara, Y. Methylglyoxal inhibits smooth muscle contraction in isolated blood vessels. J. Pharmacol. Sci. 2009, 109, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-S.; Wu, Y.; Jin, X.; Jiang, C. The SUR2B subunit of rat vascular KATP channel is targeted by miR-9a-39 induced by prolonged exposure to methylglyoxal. Am. J. Physiol. Cell Physiol. 2015, 308, C139–C145. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hall, L.M.; Kujawa, M.; Li, H.; Zhang, X.; O’Meara, M.; Ichinose, T.; Wang, J.M. Methylglyoxal triggers human aortic endothelial cell dysfunction via modulation of the KATP/MAPK pathway. Am. J. Physiol. Cell Physiol. 2019, 317, C68–C81. [Google Scholar] [CrossRef]
- Szabó, I.; Leanza, L.; Gulbins, E.; Zoratti, M. Physiology of potassium channels in the inner membrane of mitochondria. Pflüg. Arch. Eur. J. Physiol. 2012, 463, 231–246. [Google Scholar] [CrossRef]
- Szewczyk, A.; Jarmuszkiewicz, W.; Kunz, W.S. Mitochondrial potassium channels. IUBMB Life 2009, 61, 134–143. [Google Scholar] [CrossRef]
- Malinska, D.; Mirandola, S.R.; Kunz, W.S. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010, 584, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, B.; Cortassa, S.; Aon, M.A. Mitochondrial ion channels: Gatekeepers of life and death. Physiology 2005, 20, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, M.; Augustynek, B.; Kuliwak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 857, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.I.; Tisdale, M.J.; Thornalley, P.J. Glyoxalase activity during differentiation of human leakaemia cells in vitro. Leuk. Res. 1987, 11, 1141–1148. [Google Scholar] [CrossRef]
- Clelland, J.D.; Allen, R.E.; Thornalley, P.J. Inhibition of growth of human leukaemia 60 cells by S-2-hydroxyacylglutathiones and monoethyl ester derivatives. Biochem. Pharmacol. 1992, 44, 1953–1959. [Google Scholar] [CrossRef]
- Principato, G.P.; Bodo, M.; Biagioni, M.G.; Rosi, G.; Liotti, F.S. Glyoxalases and glutathione reductase activity changes in chicken liver during embryo development and after hatching. Acta Embryol. Morphol. Exp. 1982, 3, 173–179. [Google Scholar]
- Dixit, A.; Garg, L.C.; Sutrave, P.; Rao, A.R. Glyoxalase I in regenarating mouse liver exposed to carcinogens. Biochem. Intern. 1983, 7, 207–213. [Google Scholar]
- Principato, G.B.; Locci, P.; Rosi, G.; Talesa, V.; Giovannini, E. Activity changes of glyoxalases I-II and glutathione reductase in regenerating rat liver. Biochem. Intern. 1983, 6, 249–255. [Google Scholar]
- Dudani, A.K.; Srivastava, L.K.; Prasad, R. Glyoxalase-I activity and cell cycle regulation in yeast. Biochem. Biophys. Res. Commun. 1984, 119, 962–967. [Google Scholar] [CrossRef]
- Bruschelli, G.; Mariucci, G.; Principato, G.B.; Lioti, F.S. Glyoxalase activity in quiescent and proliferating human fibroblasts. Cell Mol. Biol. 1986, 32, 183–185. [Google Scholar]
- Matsuura, T.; Owada, K.; Sano, M.; Saito, S.; Tomita, I.; Ikekawa, T. Studies on methylglyoxal II/Changes of methylglyoxal level accompanying the changes of glyoxalase I and II activities in mice bearing L1210 leukemia and sarcoma 180. Chem. Pharmacol. Bull. 1986, 34, 2926–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Sethi, U.; Guha-Mukherjee, S. Induction of cell division in leaf cells of Coconut Palm by alteration of pH and its correlation with glyoxalase-I activity. J. Exp. Bot. 1988, 39, 1735–1742. [Google Scholar] [CrossRef]
- Chakravarty, T.N.; Sopory, S.K. Blue light stimulation of cell proliferation and glyoxalase I activity in callus cultures of Amaranthus paniculatus. Plant Sci. 1998, 132, 63–69. [Google Scholar] [CrossRef]
- Kalia, S.; Pal, S.; Guha-Mukherjee, S. Activation of glyoxalase I during the cell division cycle and its homology with auxin regulated genes. Plant Sci. 1998, 132, 55–62. [Google Scholar] [CrossRef]
- Gillespie, E. Concanavalin A increases glyoxalase enzyme activities in polymorphonuclear leukocytes and lymphocytes. J. Immunol. 1978, 121, 923–925. [Google Scholar]
- Gillespie, E. Effects of S-D-lactoylglutathione and inhibitors of glyoxalase I on histamine release from human leukocytes. Nature 1979, 277, 135–137. [Google Scholar] [CrossRef]
- Oliver, J.M.; Albertini, D.F.; Berlin, R.D. Effects of glutathione-oxidizing agents on microtubule assembly and microtubule-dependent surface properties of human neutrophils. J. Cell Biol. 1976, 71, 921–932. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Actin and microtubules in cell motility: Which one is in control? Traffic 2004, 5, 470–477. [Google Scholar] [CrossRef]
- Riesco, A.; Santos-Buitrago, B.; De Las Rives, J.; Knapp, M.; Santos-Garcia, G.; Talcott, C. Epidermal growth factor signaling towards proliferation: Modelling and logic interference using forward and backward search. BioMed Res. Int. 2017, 2017, 1809513. [Google Scholar] [CrossRef] [Green Version]
- Rani, R.; Kumar, S.; Sharma, A.; Mohanty, S.K.; Donnelly, B.; Tiao, G.M.; Gandhi, C.R. Mechanisms of concanavalin A-induced cytokine synthesis by hepatic stellate cells: Distinct roles of interferon regulatory factor-1 in liver injury. J. Biol. Chem. 2018, 293, 18466–18476. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J.; Greskowiak, M.; Della Bianca, V. Potentiation of secretion from neutrophils by S-D-lactoylglutathione. Med. Sci. Res. 1989, 18, 813–815. [Google Scholar]
- Allen, R.; Thornalley, P.J. The effect of S-D-lactoylglutathione on the movement of neutrophils. Biochem. Soc. Transact. 1993, 21, 161S. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. S-D-lactoylglutathione as a potential state marker for hemolysis. Med. Hypotheses 2011, 77, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, W.; Yin, L.; Du, Y.; Zhang, S.; Suo, J. Association of serum levels of deoxyribose 1-phosphate and S-lactoylglutathione with neoadjuvant chemotherapy sensitivity in patients with gastric cancer: A metabolomics study. Oncol. Lett. 2020, 19, 2231–2242. [Google Scholar] [CrossRef] [Green Version]
- Doğan, H.F.; Şenol, O.; Bolat, S.; Yıldız, S.N.; Büyüktuna, S.A.; Sarıismailoğlu, R.; Doğan, K.; Hasbek, M.; Hekim, S.N. Understanding the pathophysiological changes via untargeted metabolomics in COVID-19 patients. J. Med. Virol. 2021, 93, 2340–2349. [Google Scholar] [CrossRef]
- Kalapos, M.P. Methylglyoxal and glucose metabolism: A historical perspective and future avenues for research. Drug Metab. Drug Interact. 2008, 23, 69–91. [Google Scholar] [CrossRef]
- Kalapos, M.P. Can ageing be prevented by dietary restriction? Mech. Aging Dev. 2007, 128, 227–228. [Google Scholar] [CrossRef]
- Braun, L.; Garzó, T.; Riba, P.; Mandl, J.; Kalapos, M.P. Methylglyoxal and cell viability. Int. J. Biochem. 1994, 26, 987–990. [Google Scholar] [CrossRef]
- Karg, E.; Németh, I.; Horányi, M.; Pintér, S.; Vécsei, L.; Hollán, S. Diminished blood levels of reduced glutathione and α-tocopherol in two triose-phosphate isomerase-deficient brothers. Blood Cells Mol. Dis. 2000, 26, 91–100. [Google Scholar] [CrossRef]
- Van Wijk, R.; van Solinge, W.W. The energy-less red blood cells is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Orosz, F.; Oláh, J.; Ovádi, J. Triose-phosphate isomerase deficiency: Facts and doubts. IUBMB Life 2006, 58, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Battah, S.; Karachalias, N.; Babaei-Jadidi, R.; Horányi, M.; Baróti, K.; Hollán, S.; Thornalley, P.J. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim. Biophys. Acta 2003, 1639, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Smith, K.; Szwergold, B.S. Glyceraldehyde-3-phosphate dehydrogenase activity is an independent modifier of methylglyoxal levels in diabetes. Biochim. Biophys. Acta 2003, 1637, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Nelson, R.G.; Mauer, M.; Szwergold, B.S. α-Oxoaldehyde metabolism and diabetic complications. Biochem. Soc. Transact. 2003, 31, 1358–1363. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Drummond, K.S.; Nelson, R.G.; Howell, S.K.; Szwergold, B.S.; Mauer, M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 2005, 54, 3274–3281. [Google Scholar] [CrossRef]
- Valentine, W.N. Metabolism of human erythrocytes. Arch. Intern. Med. 1975, 138, 1307–1313. [Google Scholar] [CrossRef]
- Agar, N.S.; Board, P.G.; Bell, K. Studies of erythrocyte glyoxalase II in various domestic species: Discovery of glyoxalase II deficiency in the horse. Anim. Blood Groups Biochem. Genet. 1984, 15, 67–70. [Google Scholar] [CrossRef]
- Valentine, W.N.; Paglia, D.E.; Neerhout, R.C.; Konrad, P.N. Erythrocyte glyoxalase II with coincidental hereditary elliptocytosis. Blood 1970, 36, 797–808. [Google Scholar] [CrossRef]
- Jerzykowski, T.; Winter, R.; Matuszewski, W.; Piskorska, D. The re-evaluation of studies on the distribution of glyoxalases in animal and tumour tissues. Int. J. Biochem. 1978, 9, 853–860. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Michels, P.A.M. The metabolic repertoire of Leshmania and implications for drug discovery. In Leishmania: After the Genome; Myler, P.J., Fasel, V., Eds.; Caister Academic Press: Norfolk, UK, 2003; pp. 123–158. [Google Scholar]
- Talesa, V.N.; Ferri, I.; Bellezza, G.; Love, H.D.; Sidoni, A.; Antognelli, C. Glyoxalase 2 is involved in human prostate cancer progression as part of a mechanism driven by PTEN/P13K/AKT/mTOR signaling with involvement of PKM2 and Erα. Prostate 2017, 77, 196–210. [Google Scholar] [CrossRef]
- Ward, G.M.; Harrison, L.C. Structure of the human erythrocyte insulin receptor. Diabetes 1986, 35, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Joost, H.-G.; Bell, G.I.; Best, J.D.; Birnbaum, M.J.; Charron, M.J.; Chen, Y.T.; Doege, H.; James, D.E.; Lodish, H.F.; Moley, K.H.; et al. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E974–E976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornalley, P.J.; Jahan, I.; Ng, R. Suppression of the accumulation of triose-phosphates and increased formation of methylglyoxal in human red blood cells during hyperglycemia by thiamine in vitro. J. Biochem. 2001, 129, 543–549. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Lin, Y.-C.; Chang, G.-D. Biochemical regulation of the glyoxalase system in response to insulin signaling. Antioxidants 2021, 10, 326. [Google Scholar] [CrossRef]
- Kalapos, M.P.; Riba, P.; Garzó, T.; Mandl, J. Glucose formation from methylglyoxal in hepatocytes from streptozotocin-induced diabetic mice: The effect of insulin. Experientia 1996, 52, 827–830. [Google Scholar] [CrossRef]
- Kalapos, M.P. Where does plasma methylglyoxal originate from? Diabetes Res. Clin. Pract. 2013, 99, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Atkins, T.W.; Thornalley, P.J. Erythrocyte glyoxalase activity in genetically obese (ob/ob) and streptozotocin diabetic mice. Diabetes Res. 1989, 11, 125–129. [Google Scholar]
- Vogt-Moller, P. Ist Avitaminosis B1 eine Intoxikation mit Methylglyoxal? Biochem. Z 1929, 233, 248–250. [Google Scholar]
- Alonso, J.; Angermeyer, M.C.; Bernert, S.; Bruffaerts, R.; Brugha, T.S.; Bryson, H.; de Girolamo, G.; Graaf, R.; Demyttenaere, K.; Gasquet, I.; et al. 12-Month comorbidity patters and associated factors in Europe: Results from the European Study of the Epidemiology of Mental Disorders (ESMeD) project. Acta Psychiatr. Scand. 2004, 109 (Suppl. 420), 28–37. [Google Scholar] [CrossRef]
- Lieber, C.S. Hepatic, metabolic, and nutritional disorders of alcoholism: From pathogenesis to therapy. Crit. Rev. Clin. Lab. Sci. 2000, 37, 551–584. [Google Scholar] [CrossRef]
- Kalapos, M.P. Introduction to Alcohology; Medicina: Budapest, Hungary, 2007. [Google Scholar]
- Lonsdale, D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. eCAM 2006, 3, 49–59. [Google Scholar] [CrossRef]
- Kalapos, M.P. On the mammalian acetone metabolism/From chemistry to clinical implications. Biochim. Biophys. Acta 2003, 1621, 122–139. [Google Scholar] [CrossRef]
- Beisswenger, B.G.K.; Delucia, E.M.; Lapoint, N.; Sanford, R.J.; Beisswenger, P.J. Ketosis leads to increased methylglyoxal production on the Atkins diet. Ann. N. Y. Acad. Sci. 2005, 1043, 201–210. [Google Scholar] [CrossRef]
- Weber, Y.G.; Lerche, H. Genetics of Paroxysmal Dyskinesias. Curr. Neurol. Neurosci. Rep. 2009, 9, 206–211. [Google Scholar] [CrossRef]
- Shen, Y.; Ge, W.-P.; Li, Y.; Hirano, A.; Rohlmann, A.; Missler, M.; Tsien, R.W.; Jan, L.Y.; Fu, Y.-H.; Ptáĉek, L.J. Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc. Natl. Acad. Sci. USA 2015, 112, 2935–2941. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, N.; Vermiere, S.; Arijs, I.; Michiels, G.; Ballet, V.; Derua, R.; Waelkens, E.; Van Lommel, L.; Schuit, F.; Rutgeerts, P.; et al. Seroreactivity against glycolytic enzymes in inflammatory Bowel Disease. Inflamm. Bowel Dis. 2011, 17, 557–564. [Google Scholar] [CrossRef]
- Ritter, K.; Brestrich, H.; Nellen, B.; Kratzin, H.; Eiffer, H.; Thomssen, R. Autoantibodies against triose-phosphate isomerase/A Possible Clue to Pathogenesis of Hemolytic Anemia in Infectious Mononucleosis. J. Exp. Med. 1990, 171, 565–570. [Google Scholar] [CrossRef]
- Kültz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef]
- Yadav, S.K.; Singla-Pareek, S.L.; Ray, M.; Reddy, M.K.; Sopory, S.K. Methylglyoxal levels in plants under salinity stress are dependent on glyoxalase I and glutathione. Biochem. Biophys. Res. Commun. 2005, 337, 61–67. [Google Scholar] [CrossRef]
- Russell, J.B. Glucose toxicity in Prevotella ruminicola: Methylglyoxal accumulation and its effect on membrane physiology. Appl. Environ. Microbiol. 1993, 59, 2844–2850. [Google Scholar] [CrossRef] [Green Version]
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem.-Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of α-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Eberhardt, M.J.; Filipovic, M.R.; Leffler, A.; de la Roche, J.; Kistner, K.; Fisher, M.J.; Fleming, T.; Zimmerma, K.; Ivanovic-Burmazovic, I.; Nawroth, P.P.; et al. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1)/A possible mechanism of metabolic neuropathies. J. Biol. Chem. 2012, 287, 28291–28306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Ku, M.H.; Kang, S.-O. NAD+-linked alcohol dehydrogenase 1 regulates methylglyoxal concentration in Candida albicans. FEBS Lett. 2014, 588, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Kaur, C.; Sharma, S.; Singla-Pareek, S.L.; Sopory, S.K. Methylglyoxal detoxification in plants: Role of glyoxalase pathway. Indian J. Plant Physiol. 2016, 21, 377–390. [Google Scholar] [CrossRef]
- Fierro, C.; López-Cristolfanini, C.; Latorre, N.; Rivas, J.; Contreras-Porcia, L. Methylglyoxal metabolism in seaweeds during desiccation. Rev. Biol. Mar. Oceanogr. 2016, 51, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, B.; Goldstein, B.; Thornalley, P.J.; Rabbani, N.; Tschoepe, D. Intracellular accumulation of methylglyoxal by glyoxalase 1 knock down alters collagen homeostasis in L6 myoblasts. Int. J. Mol. Sci. 2017, 18, 480. [Google Scholar] [CrossRef]
- Inoue, Y.; Kimura, A. Glycolytic-methylglyoxal pathway Molecular evolution and stress response of glyoxalase I in Saccharomyces cerevisiae. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 1999, 75, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Espartero, J.; Sanchez-Aquayo, I.; Pardo, J.M. Molecular characterization of glyoxalase I from a higher plant: Upregulation by stress. Plant Mol. Biol. 1995, 29, 1223–1233. [Google Scholar] [CrossRef]
- Inoue, Y.; Tsujimoto, Y.; Kimura, A. Expression of the glyoxalase I gene of Saccharomyces cerevisiae is regulated by high osmolarity glycerol mitogen-activated protein kinase pathway in osmotic stress response. J. Biol. Chem. 1998, 273, 2977–2983. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, D.; Chandra, D.; Lochab, S.P.; Sarma, A.; Kale, R.K. Response of the glyoxalase system to low doses of mixed radiation. Phys. Med. 1999, 15, 27–33. [Google Scholar]
- Yadav, S.K.; Singla-Pareek, S.L.; Reddy, M.K.; Sopory, S.K. Transgenic tobacco plants overexpressing glyoxalase enzymes resist an increase methylglyoxal and maintain higher reduced glutathione levels under salinity stress. FEBS Lett. 2005, 579, 6265–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsume, Y.; Izawa, S.; Inoue, Y. Unique regulation of glyoxalase I activity during osmotic stress response in fission yeast Schizosaccharomíces pombe: Neither the mRNA nor the protein level of glyoxalase I increase under conditions that enhance its activity. Arch. Microbiol. 2003, 183, 224–227. [Google Scholar] [CrossRef]
- Hossain, M.A.; Hasanuzzaman, M.; Fujita, M. Up-regulation of antioxidant and glyoxalase systems by exogenous glycinebetaine and proline in mung bean confer tolerance to cadmium. Physiol. Mol. Biol. Plants 2010, 16, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Pharmaceuticals Containing S-lactoyl-glutathione and/or Its Salt as Active Ingredient. Japanese Patent JP2522940B2, 1988. Available online: https://patents.google.com/patent/JP2522940B2/en (accessed on 1 April 2007).
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental and clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef]
- Morcos, M.; Du, X.-H.; Pfistere, F.; Hutter, H.; Sayed, A.A.R.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalae-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef]
- Schlotterer, A.; Kukudov, G.; Bozorgmehr, F.; Hutter, H.; Du, X.-H.; Oikonomou, D.; Ibrahim, Y.; Pfisterer, F.; Rabbani, N.; Thornalley, P.; et al. C. elegans as model for the study of high glucose-mediated life span reduction. Diabetes 2009, 58, 2450–2456. [Google Scholar] [CrossRef] [Green Version]
- Scheckhuber, C.Q.; Mack, S.J.; Strobel, I.; Ricciardi, F.; Gispert, S.; Osiewacz, H.D. Modulation of the glyoxalase system in the aging model Podospora anserina: Effects on growth and lifespan. Aging 2010, 2, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Du, X.-H.; D’Agatti, V.D.; Milne, R.; Sui, G.-S.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight 2019, 4, e126154. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of glyoxalase 1 induces compensatory mechanism to achieve dicarbonyl detoxification in mammalian Schwann cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalwijk, C.G.; Steheower, C.D.A. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age related diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, F.; Zaman, M.; Thornalley, P.J.; Masters, J. Glyoxalase activities in human tumour cell lines in vitro. Anticancer Res. 1993, 13, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalapos, M.P.; Antognelli, C.; de Bari, L. Metabolic Shades of S-D-Lactoylglutathione. Antioxidants 2022, 11, 1005. https://doi.org/10.3390/antiox11051005
Kalapos MP, Antognelli C, de Bari L. Metabolic Shades of S-D-Lactoylglutathione. Antioxidants. 2022; 11(5):1005. https://doi.org/10.3390/antiox11051005
Chicago/Turabian StyleKalapos, Miklós Péter, Cinzia Antognelli, and Lidia de Bari. 2022. "Metabolic Shades of S-D-Lactoylglutathione" Antioxidants 11, no. 5: 1005. https://doi.org/10.3390/antiox11051005
APA StyleKalapos, M. P., Antognelli, C., & de Bari, L. (2022). Metabolic Shades of S-D-Lactoylglutathione. Antioxidants, 11(5), 1005. https://doi.org/10.3390/antiox11051005