
Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Protein Concentration Measurement
2.4. Detection of Free Radicals in Cells
2.5. DPPH• Free Radical Scavenging Analysis
2.6. NO Content Analysis
2.7. Western Blotting
2.8. Gelatin Zymography
2.9. Reverse Transcription and Real-Time PCR
2.10. Electroporation and Luciferase Assay
2.11. Immunofluorescence Staining
2.12. Co-Immunoprecipitation
2.13. Statistical Analysis
3. Results
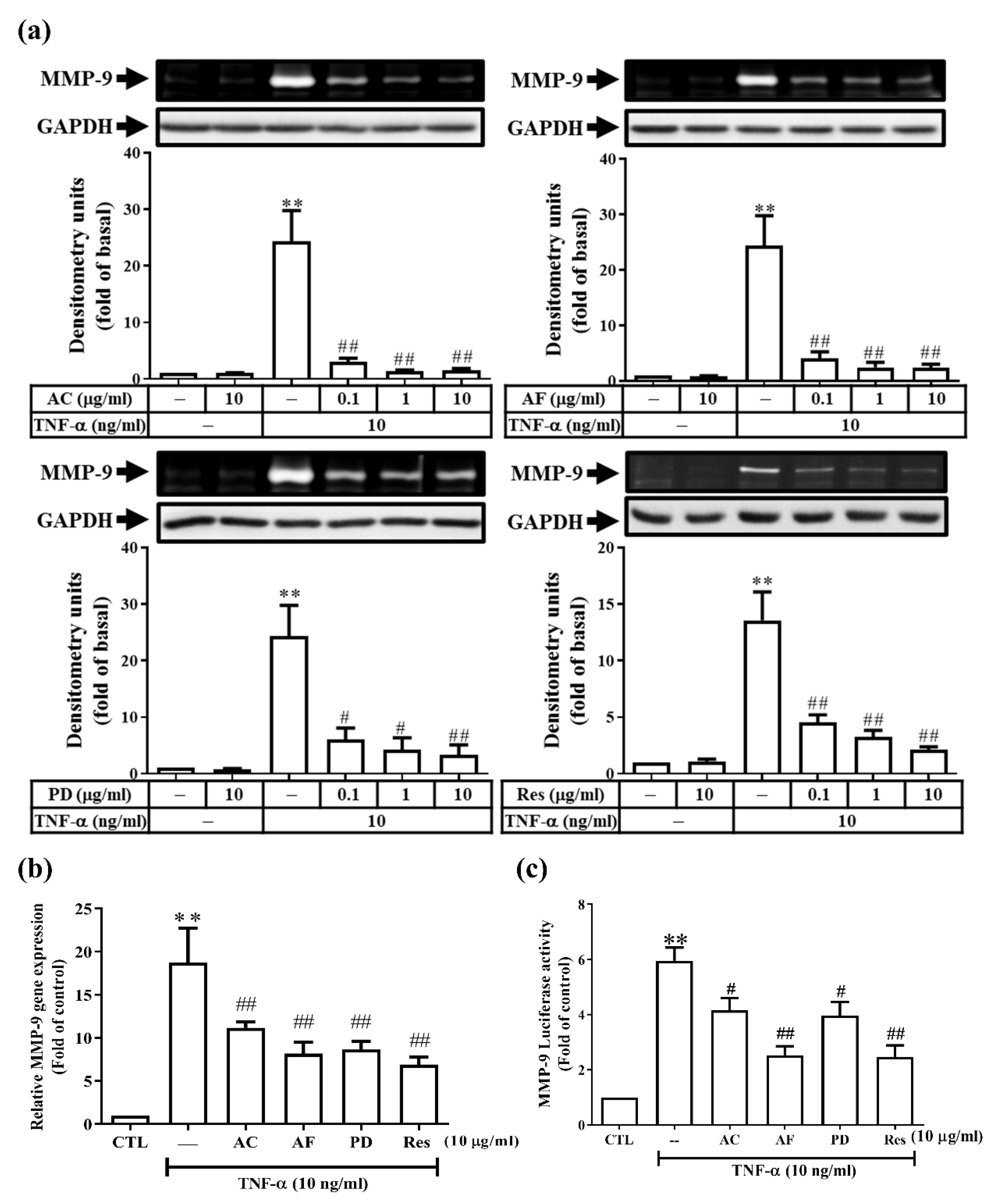
3.1. Res and Its Derivatives Attenuate TNF-α-Induced MMP-9 Expression in RMCs
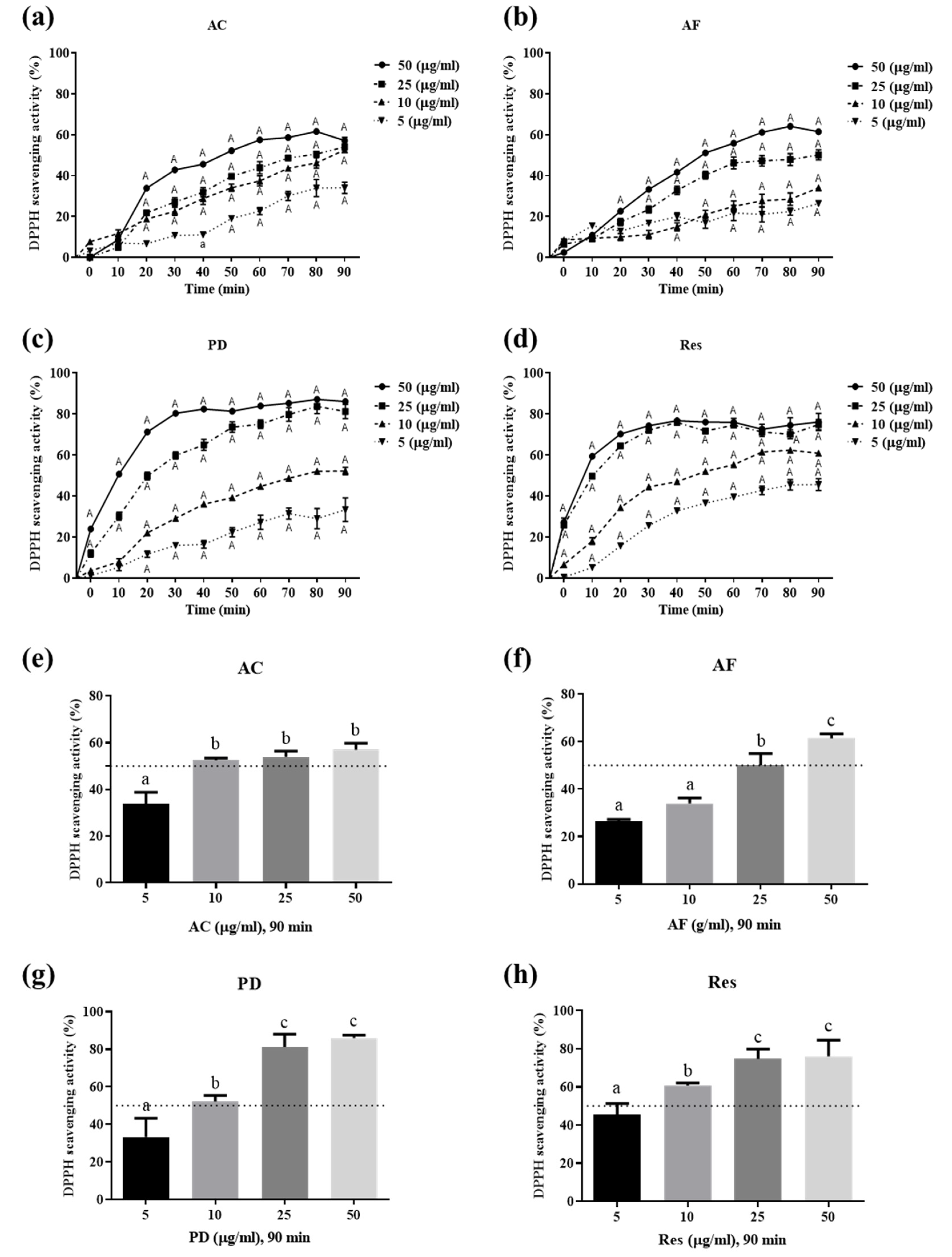
3.2. Ability of Res and Its Derivatives to Scavenge Free Radicals
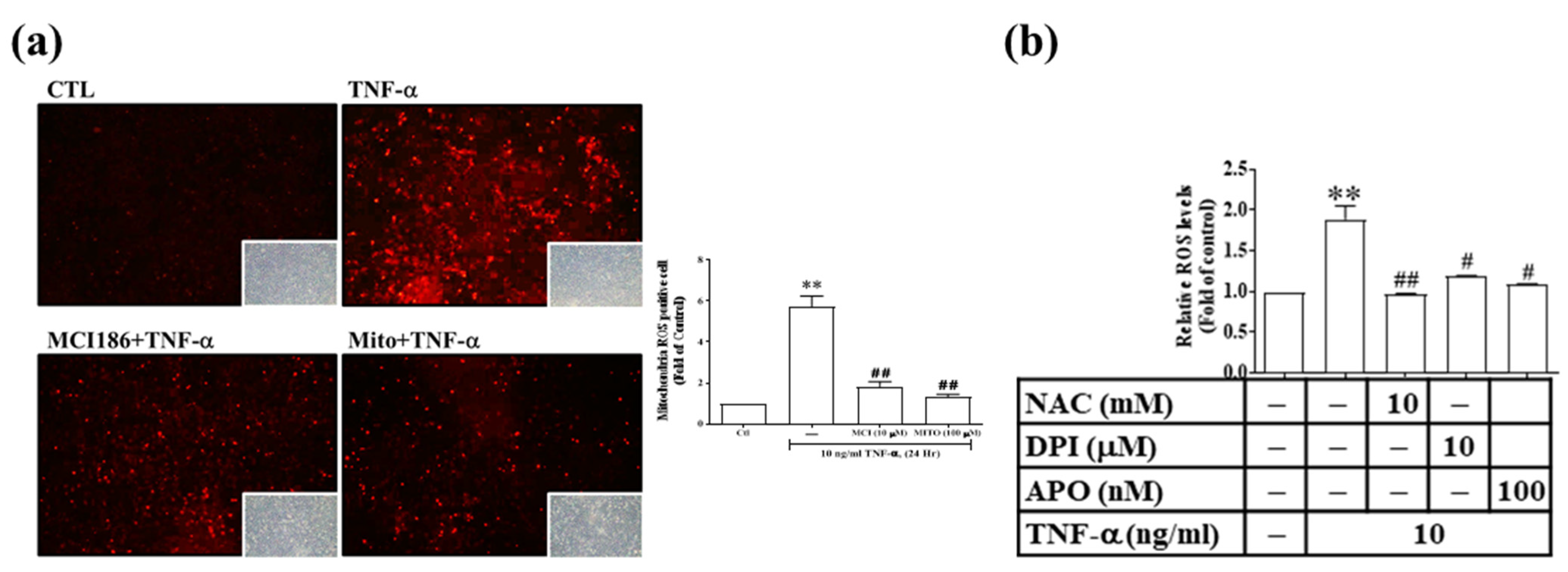
3.3. Res and Its Derivatives Relieve TNF-α-Induced Oxidative Stress in RMCs
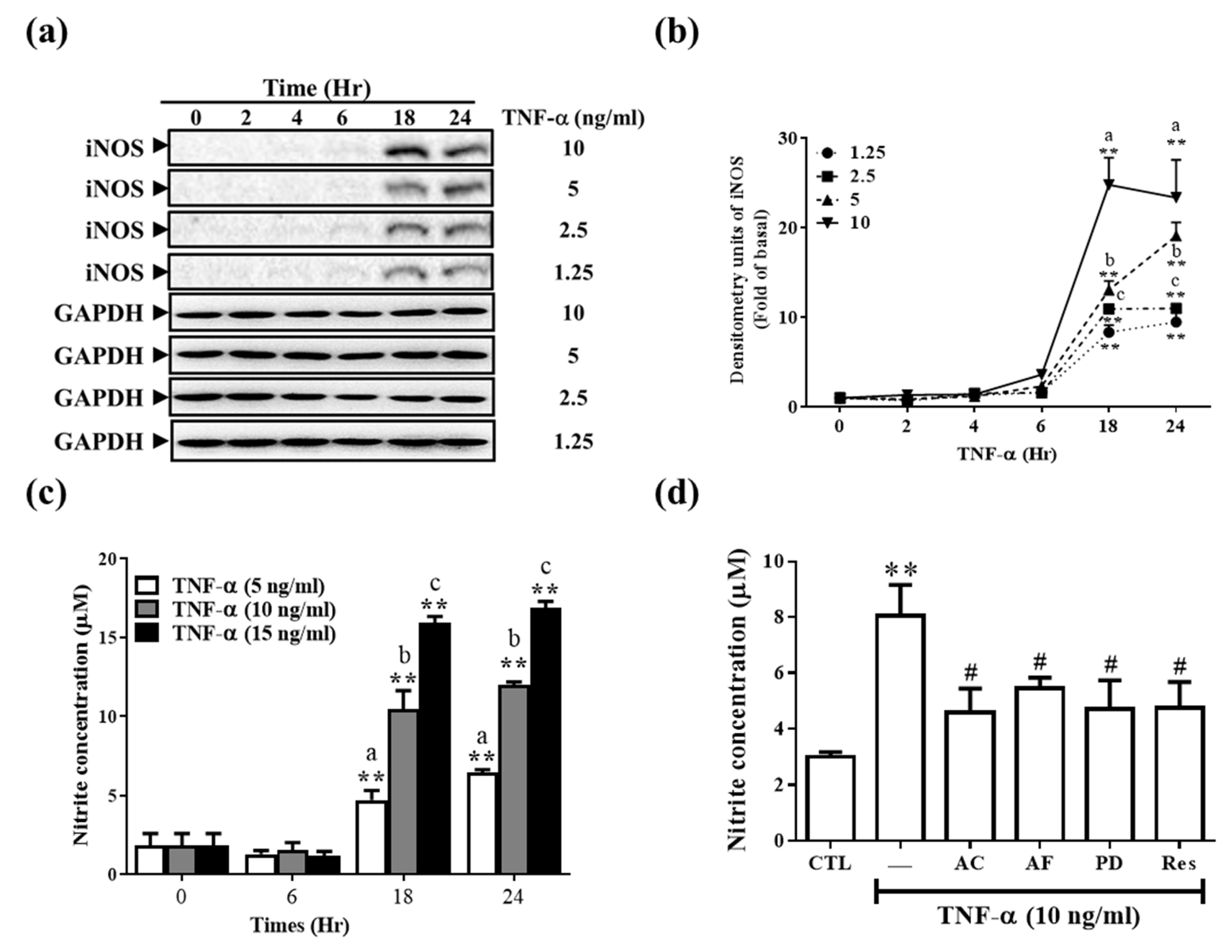
3.4. Res and Its Derivatives Relieve TNF-α-Induced iNOS Expression and NO Production in RMCs
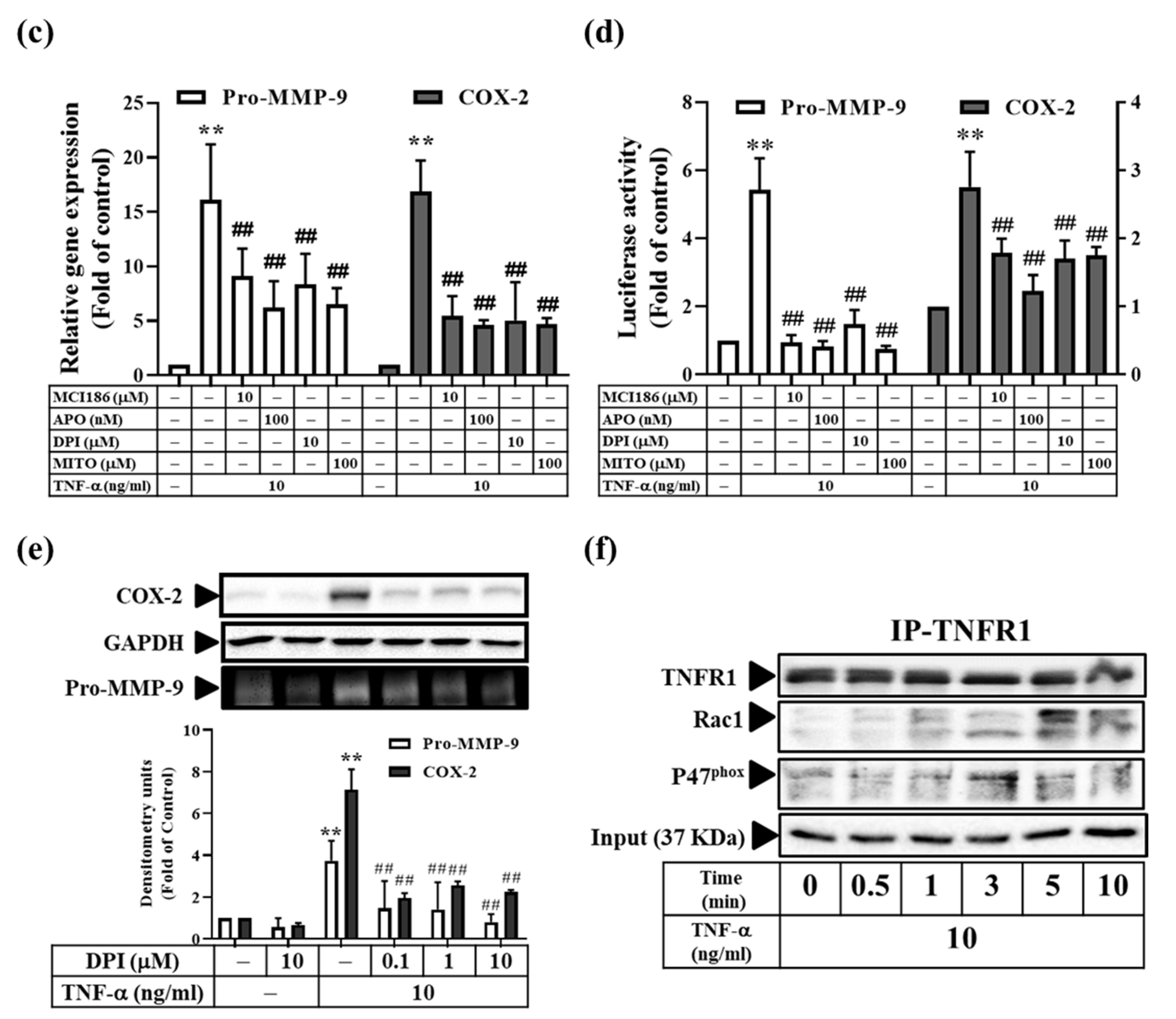
3.5. TNF-α Induces MMP-9 and COX-2 Expression via the ROS Generated from Mitochondria and NADPH Oxidase in RMCs
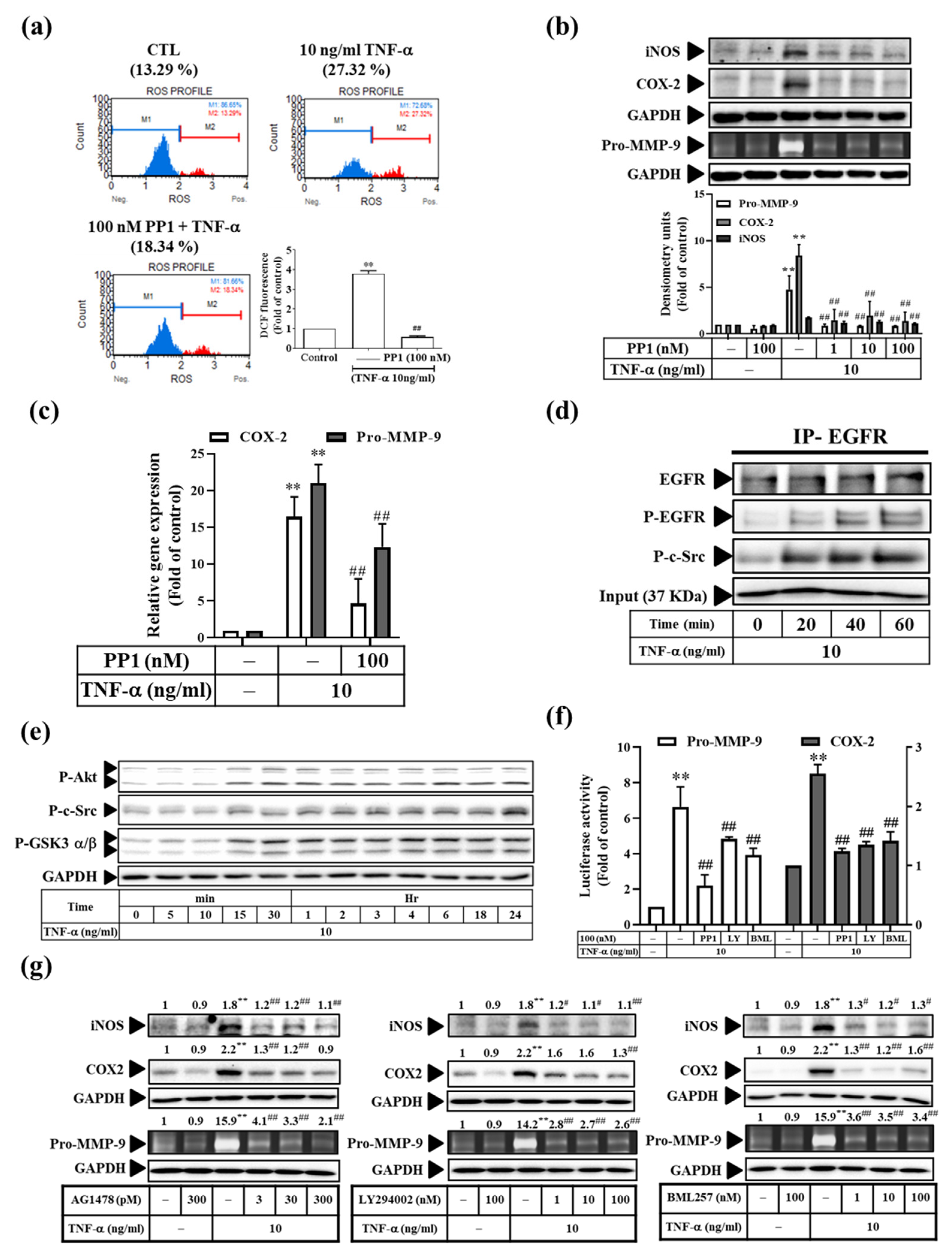
3.6. TNF-α Induces Inflammatory Protein Expression via the c-Src-Dependent EGFR Transactivation Pathway in RMCs
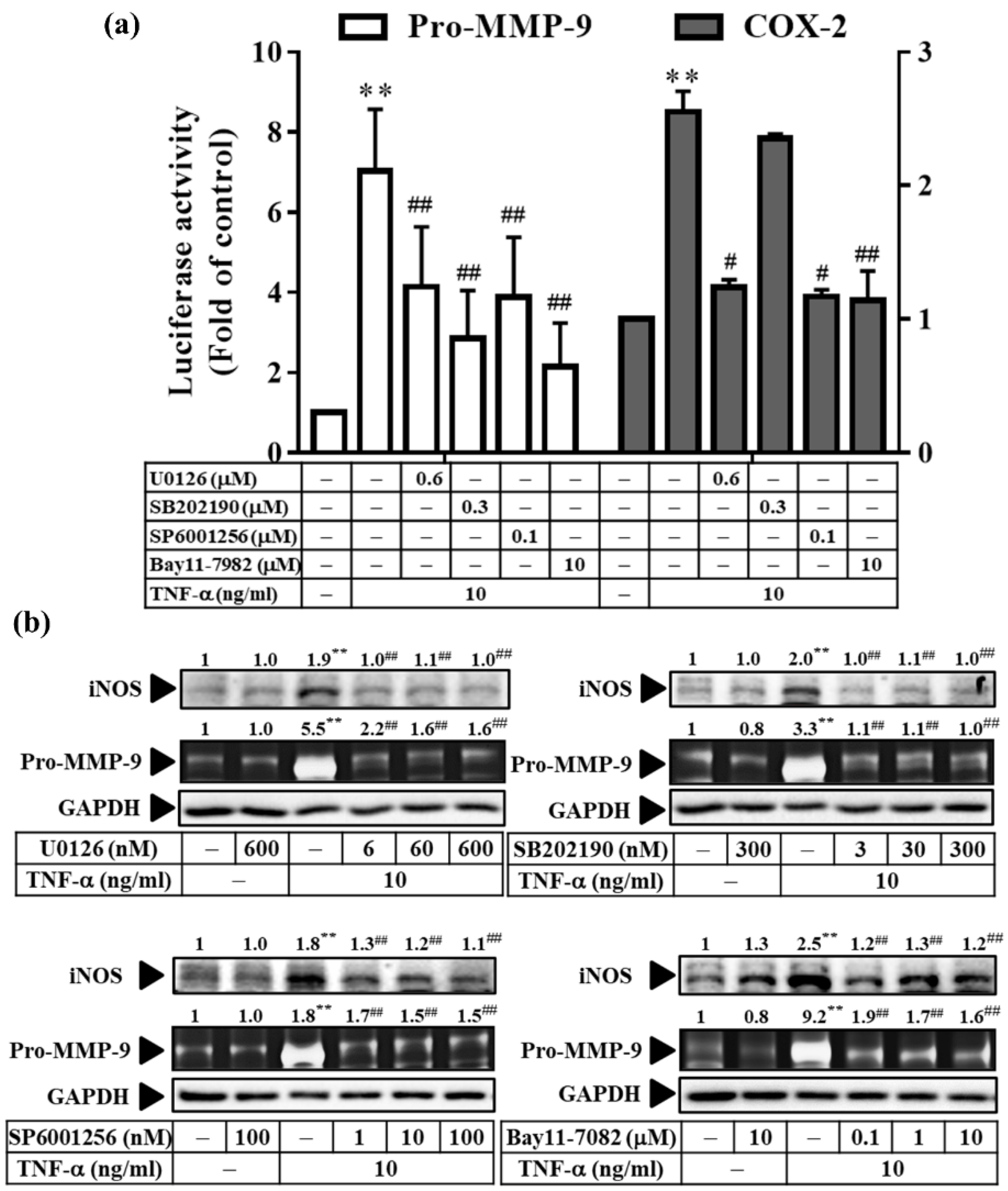
3.7. TNF-α Induces Inflammatory Protein Expression via Three Types of MAPKs and NF-κB in RMCs
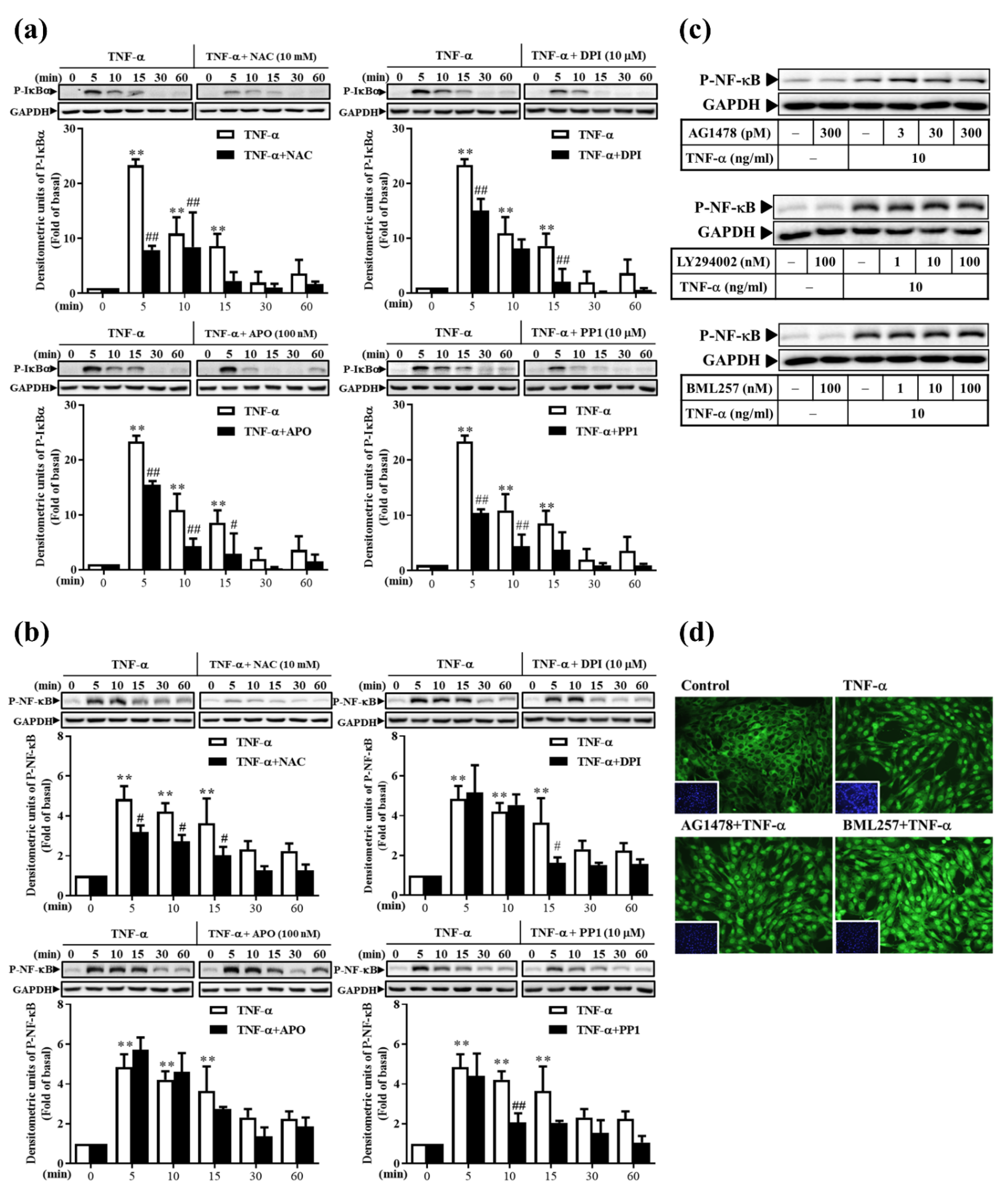
3.8. TNF-α Induces Classical NF-κB Activation via ROS, Rather Than via the EGFR Pathway, in RMCs
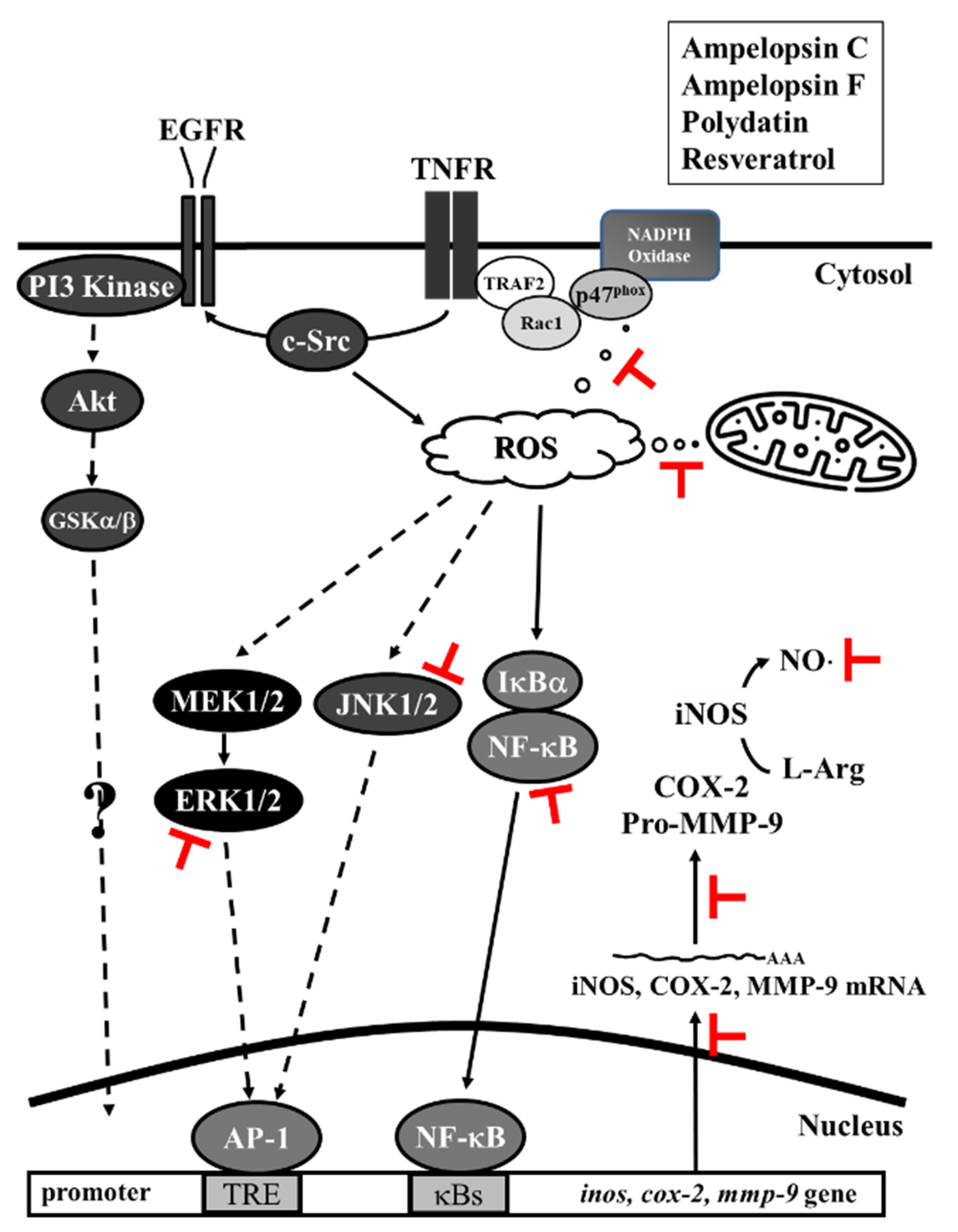
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avraham, S.; Korin, B.; Chung, J.J.; Oxburgh, L.; Shaw, A.S. The Mesangial cell—the glomerular stromal cell. Nat. Rev. Nephrol. 2021, 17, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Shi, X.; Li, J.; Jia, Y. MicroRNA-98-5p inhibits human mesangial cell proliferation and TNF-α and IL-6 secretion by targeting BTB and CNC homology 1. Exp. Ther. Med. 2021, 22, 1436. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Ren. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Limbu, M.H.; Wang, Z.; Liu, J.; Liu, L.; Zhang, X.; Chen, P.; Liu, B. MMP-2 and 9 in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, W.F.; Shen, C.C.; Chen, C.C.; Lin, C.H.; Huang, Y.L. Oligostilbenes from the roots of Vitis thunbergii. Planta Med. 2009, 75, 856–859. [Google Scholar] [CrossRef]
- Huang, Y.L.; Tsai, W.J.; Shen, C.C.; Chen, C.C. Resveratrol derivatives from the roots of Vitis thunbergii. J. Nat. Prod. 2005, 68, 217–220. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Lin, Y.K.; Yang, S.C.; Alalaiwe, A.; Lin, C.J.; Fang, J.Y.; Lin, C.F. Percutaneous absorption of resveratrol and its oligomers to relieve psoriasiform lesions: In silico, in vitro and in vivo evaluations. Int. J. Pharm. 2020, 585, 119507. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.S.; Lin, M.; Cheng, G.F. Anti-inflammatory tetramers of resveratrol from the roots of Vitis amurensis and the conformations of the seven-membered ring in some oligostilbenes. Phytochemistry 2001, 58, 357–362. [Google Scholar] [CrossRef]
- Lee, I.T.; Lin, C.F.; Huang, Y.L.; Chong, K.Y.; Hsieh, M.F.; Huang, T.H.; Cheng, C.Y. Protective mechanisms of resveratrol derivatives against TNF-α-induced inflammatory responses in rat mesangial cells. Cytokine 2019, 113, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Nassiri-Asl, M.; Hosseinzadeh, H. Review of the pharmacological effects of Vitis vinifera (Grape) and its bioactive compounds. Phytother. Res. PTR 2009, 23, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yan, X. From resveratrol to its derivatives: New sources of natural antioxidant. Curr. Med. Chem. 2013, 20, 1005–1017. [Google Scholar]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutiérrez-Juárez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [Green Version]
- Frankel, E.N.; Waterhouse, A.L.; Kinsella, J.E. Inhibition of human LDL oxidation by resveratrol. Lancet 1993, 341, 1103–1104. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Pezzuto, J.M.; Kondratyuk, T.P.; Ogas, T. Resveratrol derivatives: A patent review (2009–2012). Expert Opin. Ther. Pat. 2013, 23, 1529–1546. [Google Scholar] [CrossRef]
- Keylor, M.H.; Matsuura, B.S.; Stephenson, C.R. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. [Google Scholar] [CrossRef]
- Flamini, R.; Mattivi, F.; De Rosso, M.; Arapitsas, P.; Bavaresco, L. Advanced knowledge of three important classes of grape phenolics: Anthocyanins, stilbenes and flavonols. Int. J. Mol. Sci. 2013, 14, 19651–19669. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, T.; Saiko, P.; Fritzer-Szekeres, M.; Djavan, B.; Jäger, W. Chemopreventive effects of resveratrol and resveratrol derivatives. Ann. N. Y. Acad. Sci. 2011, 1215, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Roberti, M.; Raul, F.; Pendurthi, U.R. Suppression of human monocyte tissue factor induction by red wine phenolics and synthetic derivatives of resveratrol. Thromb. Res. 2007, 119, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zeng, Z.; Li, T.; Xu, S.; Wang, X.; Chen, Z.; Lin, C. Polydatin Inhibits Mitochondrial Dysfunction in the Renal Tubular Epithelial Cells of a Rat Model of Sepsis-Induced Acute Kidney Injury. Anesth. Analg. 2015, 121, 1251–1260. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Haque, A.; Hsieh, M.F.; Imran Hassan, S.; Faizi, M.S.H.; Dege, N.; Khan, M.S. 1,4-Disubstituted 1H-1,2,3-Triazoles for Renal Diseases: Studies of Viability, Anti-Inflammatory, and Antioxidant Activities. Int. J. Mol. Sci. 2020, 21, 3823. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lin, C.C.; Lee, C.Y.; Hsieh, P.W.; Yang, C.M. Protective effects of (-)-epigallocatechin-3-gallate against TNF-α-induced lung inflammation via ROS-dependent ICAM-1 inhibition. J. Nutr. Biochem. 2013, 24, 124–136. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Kuo, C.T.; Lin, C.C.; Hsieh, H.L.; Yang, C.M. IL-1beta induces expression of matrix metalloproteinase-9 and cell migration via a c-Src-dependent, growth factor receptor transactivation in A549 cells. Br. J. Pharmacol. 2010, 160, 1595–1610. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef]
- Bian, X.; Bai, Y.; Su, X.; Zhao, G.; Sun, G.; Li, D. Knockdown of periostin attenuates 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, and inflammation in rats. J. Cell. Physiol. 2019, 234, 22857–22873. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Uddin, M.J.; Kim, E.H.; Hannan, M.A.; Ha, H. Pharmacotherapy against Oxidative Stress in Chronic Kidney Disease: Promising Small Molecule Natural Products Targeting Nrf2-HO-1 Signaling. Antioxidants 2021, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Quoilin, C.; Mouithys-Mickalad, A.; Lécart, S.; Fontaine-Aupart, M.P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Et Biophys. Acta 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 3), S241–S245. [Google Scholar] [CrossRef] [Green Version]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Swärd, P.; Rippe, B. Acute and sustained actions of hyperglycaemia on endothelial and glomerular barrier permeability. Acta Physiol. 2012, 204, 294–307. [Google Scholar] [CrossRef]
- Xu, Y.; Ruan, S.; Wu, X.; Chen, H.; Zheng, K.; Fu, B. Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. Int. J. Mol. Med. 2013, 31, 628–636. [Google Scholar] [CrossRef]
- Wang, J.N.; Yang, Q.; Yang, C.; Cai, Y.T.; Xing, T.; Gao, L.; Wang, F.; Chen, X.; Liu, X.Q.; He, X.Y.; et al. Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 2020, 32, 101479. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Hahn, S.A.; Miller, M.P.; Sanker, S.; Calderon, M.J.; Sullivan, M.; Dosunmu-Ogunbi, A.M.; Fazzari, M.; Li, Y.; Reynolds, M.; et al. Cooperation between CYB5R3 and NOX4 via coenzyme Q mitigates endothelial inflammation. Redox Biol. 2021, 47, 102166. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Gluschko, A.; Wiegmann, K.; Farid, A.; Wolf, A.; Utermöhlen, O.; Krut, O.; Krönke, M.; Schramm, M. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of NEMO. Sci. Signal. 2019, 12, eaar5926. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef] [Green Version]
- Geiszt, M.; Lekstrom, K.; Witta, J.; Leto, T.L. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J. Biol. Chem. 2003, 278, 20006–20012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeya, R.; Ueno, N.; Kami, K.; Taura, M.; Kohjima, M.; Izaki, T.; Nunoi, H.; Sumimoto, H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J. Biol. Chem. 2003, 278, 25234–25246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar] [CrossRef]
- Martano, M.; Stiuso, P.; Facchiano, A.; De Maria, S.; Vanacore, D.; Restucci, B.; Rubini, C.; Caraglia, M.; Ravagnan, G.; Lo Muzio, L. Aryl hydrocarbon receptor, a tumor grade-associated marker of oral cancer, is directly downregulated by polydatin: A pilot study. Oncol. Rep. 2018, 40, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayyaz, S.; Qureshi, M.Z.; Alhewairini, S.S.; Avnioglu, S.; Attar, R.; Sabitaliyevich, U.Y.; Buha, A.; Salahuddin, H.; Adylova, A.; Tahir, F.; et al. Regulation of signaling pathways by Ampelopsin (Dihydromyricetin) in different cancers: Exploring the highways and byways less travelled. Cell. Mol. Biol. 2019, 65, 15–20. [Google Scholar] [CrossRef]
- Chang, Y.; Yuan, L.; Liu, J.; Muhammad, I.; Cao, C.; Shi, C.; Zhang, Y.; Li, R.; Li, C.; Liu, F. Dihydromyricetin attenuates Escherichia coli lipopolysaccharide-induced ileum injury in chickens by inhibiting NLRP3 inflammasome and TLR4/NF-κB signalling pathway. Vet. Res. 2020, 51, 72. [Google Scholar] [CrossRef]
- Hou, L.; Jiang, F.; Huang, B.; Zheng, W.; Jiang, Y.; Cai, G.; Liu, D.; Hu, C.Y.; Wang, C. Dihydromyricetin resists inflammation-induced muscle atrophy via ryanodine receptor-CaMKK-AMPK signal pathway. J. Cell. Mol. Med. 2021, 25, 9953–9971. [Google Scholar] [CrossRef]
- Zou, J.; Xu, M.X.; Li, F.; Wang, Y.H.; Li, X.Q.; Yu, D.J.; Ma, Y.J.; Zhang, Y.Y.; Sun, X.D. Icaritin alleviates docetaxel-induced skin injury by suppressing reactive oxygen species via estrogen receptors. Thorac. Cancer 2022, 13, 190–201. [Google Scholar] [CrossRef]
- Kim, H.R.; Kang, S.Y.; Kim, H.O.; Park, C.W.; Chung, B.Y. Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation. Int. J. Mol. Sci. 2020, 21, 2195. [Google Scholar] [CrossRef] [Green Version]
- Degovics, D.; Hartmann, P.; Németh, I.B.; Árva-Nagy, N.; Kaszonyi, E.; Szél, E.; Strifler, G.; Bende, B.; Krenács, L.; Kemény, L.; et al. A novel target for the promotion of dermal wound healing: Ryanodine receptors. Toxicol. Appl. Pharmacol. 2019, 366, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ke, J.; Wu, X.; Yan, Y. Astragaloside prevents UV-induced keratinocyte injury by regulating TLR4/NF-κB pathway. J. Cosmet. Dermatol. 2022, 21, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Chen, C.C.; Shen, C.C.; Chui, C.H.; Huang, Y.L. Simultaneous determination of resveratrol derivatives in Vitis thunbergii plant by high performance liquid chromatography. J. Food Drug Anal. 2012, 20, 495–500. [Google Scholar]
- Meneses-Gutiérrez, C.L.; Hernández-Damián, J.; Pedraza-Chaverri, J.; Guerrero-Legarreta, I.; Téllez, D.I.; Jaramillo-Flores, M.E. Antioxidant Capacity and Cytotoxic Effects of Catechins and Resveratrol Oligomers Produced by Enzymatic Oxidation against T24 Human Urinary Bladder Cancer Cells. Antioxidants 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, H.Y.; Liu, Z.M.; Huang, H.S. NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes. Arch. Toxicol. 2012, 86, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Hossain, E.; Anand-Srivastava, M.B. Resveratrol prevents angiotensin II-induced hypertrophy of vascular smooth muscle cells through the transactivation of growth factor receptors. Can. J. Physiol. Pharmacol. 2017, 95, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.-T.; Lin, H.-C.; Huang, T.-H.; Tseng, C.-N.; Cheng, H.-T.; Huang, W.-C.; Cheng, C.-Y. Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells. Antioxidants 2022, 11, 835. https://doi.org/10.3390/antiox11050835
Lee I-T, Lin H-C, Huang T-H, Tseng C-N, Cheng H-T, Huang W-C, Cheng C-Y. Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells. Antioxidants. 2022; 11(5):835. https://doi.org/10.3390/antiox11050835
Chicago/Turabian StyleLee, I-Ta, Horng-Chyuan Lin, Tse-Hung Huang, Chi-Nan Tseng, Hao-Tsa Cheng, Wen-Chung Huang, and Ching-Yi Cheng. 2022. "Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells" Antioxidants 11, no. 5: 835. https://doi.org/10.3390/antiox11050835
APA StyleLee, I. -T., Lin, H. -C., Huang, T. -H., Tseng, C. -N., Cheng, H. -T., Huang, W. -C., & Cheng, C. -Y. (2022). Anti-Inflammatory Effect of Resveratrol Derivatives via the Downregulation of Oxidative-Stress-Dependent and c-Src Transactivation EGFR Pathways on Rat Mesangial Cells. Antioxidants, 11(5), 835. https://doi.org/10.3390/antiox11050835