
Crosstalk between Heme Oxygenase-1 and Iron Metabolism in Macrophages: Implications for the Modulation of Inflammation and Immunity



{kind=link}
{kind=link}
Abstract
:1. Introduction
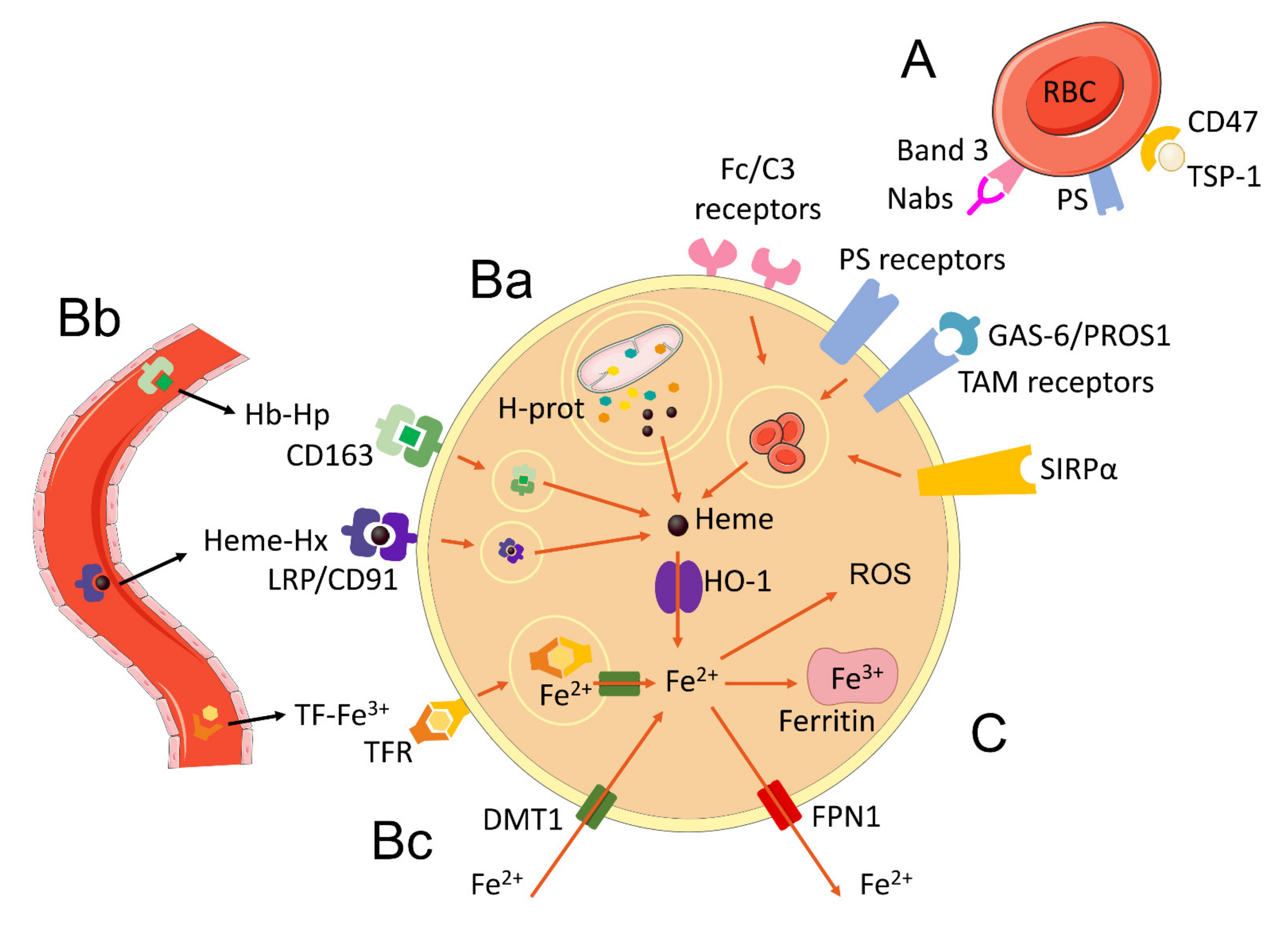
2. Heme Acquisition by Macrophages
2.1. Erythrophagocytosis
2.2. Haptoglobin and Hemopexin
2.3. Autophagy of Hemoproteins and Mitophagy
3. Heme Degradation by HO-1 and Iron Release
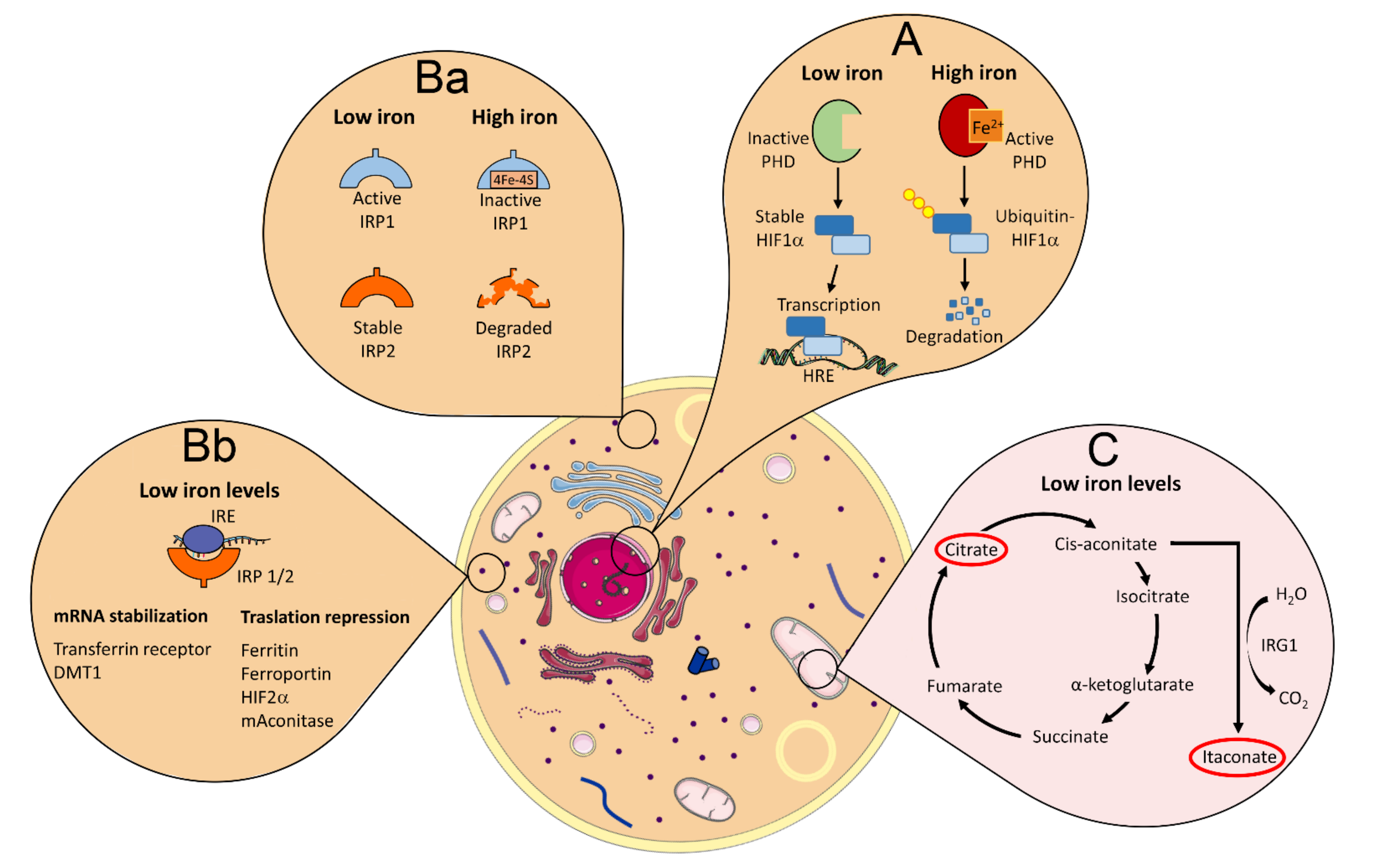
4. Cross Regulation of Iron Homeostasis, Inflammation, and Immunity
4.1. Iron Regulation of HIF1α Expression
4.2. Iron Regulation of IRPs/IRE Interactions
4.3. Iron Induction of ROS Generation by Fenton Reaction
4.4. Iron and Polarization of M1 and M2 Macrophages
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Soares, M.P.; Bach, F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, G.; Yoshida, T. Heme degradation by the microsomal heme oxygenase system. Trends Biochem. Sci. 1980, 5, 323–325. [Google Scholar] [CrossRef]
- Costa, D.L.; Amaral, E.P.; Andrade, B.B.; Sher, A. Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens. Antioxidants 2020, 9, 1205. [Google Scholar] [CrossRef] [PubMed]
- Choby, J.E.; Skaar, E.P. Heme Synthesis and Acquisition in Bacterial Pathogens. J. Mol. Biol. 2016, 428, 3408–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piel, R.B., III; Dailey, H.A., Jr.; Medlock, A.E. The mitochondrial heme metabolon: Insights into the complex(ity) of heme synthesis and distribution. Mol. Genet. Metab. 2019, 128, 198–203. [Google Scholar] [CrossRef]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. 2019, 10, 72. [Google Scholar] [CrossRef]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef]
- Han, D.; Gao, J.; Gu, X.; Hengstler, J.G.; Zhang, L.; Shahid, M.; Ali, T.; Han, B. P21(Waf1/Cip1) depletion promotes dexamethasone-induced apoptosis in osteoblastic MC3T3-E1 cells by inhibiting the Nrf2/HO-1 pathway. Arch. Toxicol. 2018, 92, 679–692. [Google Scholar] [CrossRef]
- Ji, Y.; Yin, W.; Liang, Y.; Sun, L.; Yin, Y.; Zhang, W. Anti-Inflammatory and Anti-Oxidative Activity of Indole-3-Acetic Acid Involves Induction of HO-1 and Neutralization of Free Radicals in RAW264.7 Cells. Int. J. Mol. Sci. 2020, 21, 1579. [Google Scholar] [CrossRef] [Green Version]
- Duckers, H.J.; Boehm, M.; True, A.L.; Yet, S.F.; San, H.; Park, J.L.; Webb, R.C.; Lee, M.E.; Nabel, G.J.; Nabel, E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001, 7, 693–698. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Wagener, F.A.; Yachie, A.; Wiegerinck, E.T.; Kemna, E.H.; Swinkels, D.W. Hepcidin suppression and defective iron recycling account for dysregulation of iron homeostasis in heme oxygenase-1 deficiency. J. Cell. Mol. Med. 2009, 13, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.S.; Chau, L.Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef]
- Espinoza, J.A.; Gonzalez, P.A.; Kalergis, A.M. Modulation of Antiviral Immunity by Heme Oxygenase-1. Am. J. Pathol. 2017, 187, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Lamb, N.J.; Quinlan, G.J.; Mumby, S.; Evans, T.W.; Gutteridge, J.M. Haem oxygenase shows pro-oxidant activity in microsomal and cellular systems: Implications for the release of low-molecular-mass iron. Biochem. J. 1999, 344, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klei, T.R.L.; Meinderts, S.M.; Berg, T.K.v.d.; Bruggen, R.v. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Arese, P.; Turrini, F.; Schwarzer, E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell. Physiol. Biochem. 2005, 16, 133–146. [Google Scholar] [CrossRef]
- Lutz, H.U.; Bussolino, F.; Flepp, S.F.R.; Stammler, P.; Kazatchkine, M.D.; Arese, P. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc. Natl. Acad. Sci. USA 1987, 84, 7368–7372. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Jilani, K.; Lang, E. Therapeutic potential of manipulating suicidal erythrocyte death. Expert Opin. Ther. Targets 2015, 19, 1219–1227. [Google Scholar] [CrossRef]
- Fernandez-Boyanapalli, R.F.; Frasch, S.C.; McPhillips, K.; Vandivier, R.W.; Harry, B.L.; Riches, D.W.H.; Henson, P.M.; Bratton, D.L. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 2009, 113, 2047–2055. [Google Scholar] [CrossRef] [Green Version]
- Raymond, A.; Ensslin, M.A.; Shur, B.D. SED1/MFG-E8: A bi-motif protein that orchestrates diverse cellular interactions. J. Cell. Biochem. 2009, 106, 957–966. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Karisola, P.; Peña-Cruz, V.; Dorfman, D.M.; Jinushi, M.; Umetsu, S.E.; Butte, M.J.; Nagumo, H.; Chernova, I.; Zhu, B.; et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 2007, 27, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Jung, M.-Y.; Kim, H.-J.; Lee, S.-J.; Kim, S.-Y.; Lee, B.-H.; Kwon, T.-H.; Park, R.-W.; Kim, I.-S. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008, 15, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Tian, L.; Voss, O.H.; Margulies, D.H.; Krzewski, K.; Coligan, J.E. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ. 2014, 21, 1746–1757. [Google Scholar] [CrossRef] [Green Version]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Burger, P.; Hilarius-Stokman, P.; Korte, D.d.; Berg, T.K.v.d.; Bruggen, R.v. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 2012, 119, 5512–5521. [Google Scholar] [CrossRef] [Green Version]
- Vogt, A.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [Green Version]
- Belcher, J.D.; Nath, K.A.; Vercellotti, G.M. Vasculotoxic and Proinflammatory Effects of Plasma Heme: Cell Signaling and Cytoprotective Responses. ISRN Oxidative Med. 2013, 2013, 831596. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.L.; Champion, H.C.; Campbell-Lee, S.A.; Bivalacqua, T.J.; Manci, E.A.; Diwan, B.A.; Schimel, D.M.; Cochard, A.E.; Wang, X.; Schechter, A.N.; et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007, 109, 3088–3098. [Google Scholar] [CrossRef] [PubMed]
- Windsant, I.C.V.; Wit, N.C.J.d.; Sertorio, J.T.C.; Bijnen, A.A.v.; Ganushchak, Y.M.; Heijmans, J.H.; Tanus-Santos, J.E.; Jacobs, M.J.; Maessen, J.G.; Buurman, W.A. Hemolysis during cardiac surgery is associated with increased intravascular nitric oxide consumption and perioperative kidney and intestinal tissue damage. Front. Physiol. 2014, 5, 340. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Heiss, C.; Drexhage, C.; Kehmeier, E.S.; Balzer, J.; Mühlfeld, A.; Merx, M.W.; Lauer, T.; Kühl, H.; Floege, J.; et al. Hemodialysis-induced release of hemoglobin limits nitric oxide bioavailability and impairs vascular function. J. Am. Coll. Cardiol. 2010, 55, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Meegan, J.E.; Bastarache, J.A.; Ware, L.B. Toxic effects of cell-free hemoglobin on the microvascular endothelium: Implications for pulmonary and nonpulmonary organ dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L429–L439. [Google Scholar] [CrossRef]
- Barber, B.E.; Grigg, M.J.; Piera, K.A.; William, T.; Cooper, D.J.; Plewes, K.; Dondorp, A.M.; Yeo, T.W.; Anstey, N.M. Intravascular haemolysis in severe Plasmodium knowlesi malaria: Association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerg. Microbes Infect. 2018, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Atichartakarn, V.; Chuncharunee, S.; Archararit, N.; Udomsubpayakul, U.; Aryurachai, K. Intravascular hemolysis, vascular endothelial cell activation and thrombophilia in splenectomized patients with hemoglobin E/β-thalassemia disease. Acta Haematol. 2014, 132, 100–107. [Google Scholar] [CrossRef]
- Posta, N.; Csősz, É.; Oros, M.; Pethő, D.; Potor, L.; Kalló, G.; Hendrik, Z.; Sikura, K.É.; Méhes, G.; Tóth, C.; et al. Hemoglobin oxidation generates globin-derived peptides in atherosclerotic lesions and intraventricular hemorrhage of the brain, provoking endothelial dysfunction. Lab. Investig. 2020, 100, 986–1002. [Google Scholar] [CrossRef]
- Shaver, C.M.; Wickersham, N.; McNeil, J.B.; Nagata, H.; Miller, A.; Landstreet, S.R.; Kuck, J.L.; Diamond, J.M.; Lederer, D.J.; Kawut, S.M.; et al. Cell-free hemoglobin promotes primary graft dysfunction through oxidative lung endothelial injury. JCI Insight 2018, 3, e98546. [Google Scholar] [CrossRef] [Green Version]
- Loomis, Z.; Eigenberger, P.; Redinius, K.; Lisk, C.; Karoor, V.; Nozik-Grayck, E.; Ferguson, S.K.; Hassell, K.; Nuss, R.; Stenmark, K.; et al. Hemoglobin induced cell trauma indirectly influences endothelial TLR9 activity resulting in pulmonary vascular smooth muscle cell activation. PLoS ONE 2017, 12, e0171219. [Google Scholar] [CrossRef]
- Vercellotti, G.M.; Zhang, P.; Nguyen, J.; Abdulla, F.; Chen, C.; Nguyen, P.; Nowotny, C.; Steer, C.J.; Smith, A.; Belcher, J.D. Hepatic Overexpression of Hemopexin Inhibits Inflammation and Vascular Stasis in Murine Models of Sickle Cell Disease. Mol. Med. 2016, 22, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Buehler, P.W.; Humar, R.; Schaer, D.J. Haptoglobin Therapeutics and Compartmentalization of Cell-Free Hemoglobin Toxicity. Trends Mol. Med. 2020, 26, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Paoli, M.; Anderson, B.F.; Baker, H.M.; Morgan, W.T.; Smith, A.; Baker, E.N. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat. Struct. Biol. 1999, 6, 926–931. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Møller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Moestrup, S.K. Receptor targeting of hemoglobin mediated by the haptoglobins: Roles beyond heme scavenging. Blood 2009, 114, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Masi, A.d.; Simone, G.D.; Ciaccio, C.; D’Orso, S.; Coletta, M.; Ascenzi, P. Haptoglobin: From hemoglobin scavenging to human health. Mol. Aspects Med. 2020, 73, 100851. [Google Scholar] [CrossRef]
- Buehler, P.W.; Abraham, B.; Vallelian, F.; Linnemayr, C.; Pereira, C.P.; Cipollo, J.F.; Jia, Y.; Mikolajczyk, M.; Boretti, F.S.; Schoedon, G.; et al. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 2009, 113, 2578–2586. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Schaer, D.J.; Schaer, C.A.; Buehler, P.W.; Boykins, R.A.; Schoedon, G.; Alayash, A.I.; Schaffner, A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 2006, 107, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Moestrup, S.K.; Gliemann, J.; Pallesen, G. Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 1992, 269, 375–382. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Hojrup, P.; Moller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Smith, L.J.; Kahraman, A.; Thornton, J.M. Heme proteins--diversity in structural characteristics, function, and folding. Proteins 2010, 78, 2349–2368. [Google Scholar] [CrossRef]
- Li, T.; Bonkovsky, H.L.; Guo, J.-t. Structural analysis of heme proteins: Implications for design and prediction. BMC Struct. Biol. 2011, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Reedy, C.J.; Gibney, B.R. Heme protein assemblies. Chem. Rev. 2004, 104, 617–649. [Google Scholar] [CrossRef]
- Shimizu, T.; Lengalova, A.; Martínek, V.; Martínková, M. Heme: Emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 2019, 48, 5624–5657. [Google Scholar] [CrossRef]
- Gebicka, L. Redox reactions of heme proteins with flavonoids. J. Inorg. Biochem. 2020, 208, 111095. [Google Scholar] [CrossRef]
- Henry, Y.; Guissani, A. Interactions of nitric oxide with hemoproteins: Roles of nitric oxide in mitochondria. Cell. Mol. Life Sci. 1999, 55, 1003–1014. [Google Scholar] [CrossRef]
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2019, 133, 88–100. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Srisook, K.; Kim, C.; Cha, Y.-N. Molecular mechanisms involved in enhancing HO-1 expression: De-repression by heme and activation by Nrf2, the “one-two” punch. Antioxid. Redox Signal. 2005, 7, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Renal Physiol. 2003, 284, F743–F752. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Canesin, G.; Hejazi, S.M.; Swanson, K.D.; Wegiel, B. Heme-Derived Metabolic Signals Dictate Immune Responses. Front. Immunol. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Kuang, Y.; Wang, Q. Iron and lung cancer. Cancer Lett. 2019, 464, 56–61. [Google Scholar] [CrossRef]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antiox. Redox Signal. 2014, 20, 1754–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Eid, C.; Hemadi, M.; Ha-Duong, N.T.; El Hage Chahine, J.M. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta 2014, 1840, 1771–1781. [Google Scholar] [CrossRef]
- Gomme, P.T.; McCann, K.B.; Bertolini, J. Transferrin: Structure, function and potential therapeutic actions. Drug Discov. Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philpott, C.C.; Jadhav, S. The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 2019, 133, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Tabuchi, M.; Kawai, Y.; Yasui, Y.; Akagi, R.; Kishi, F. Heme and non-heme iron transporters in non-polarized and polarized cells. BMC Cell. Biol. 2010, 11, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Nat. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Path. 2015, 11, e1004998. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Iron and infection. Int. J. Hematol. 2018, 107, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Gaël, N.; Caroline, C.; Lydie, V.; Jean Louis, D.; Xavier, B.; Isabelle, D.; Carole, B.; Axel, K.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.-P.; Townsend, A.R.M.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Annelies, S.; Zinkernagel; Datta, V.; Xavier Lauth, R.S.J.; Nizet, V. TLR4–dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Abreu, R.; Quinn, F.; Giri, P.K. Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. 2018, 2, 1089–1100. [Google Scholar] [CrossRef]
- Renassiaa, C.; Peyssonnaux, C. New insights into the links between hypoxia and iron homeostasis. Curr. Opin. Hematol. 2019, 26, 125–130. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Res. Com. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibasaki, F. Hypoxia-Inducible Factor as an Angiogenic Master Switch. Front. Pediatr. 2015, 3, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wiesener, M.S.; Turley, H.; Allen, W.E.; Willam, C.; Eckardt, K.U.; Talks, K.L.; Wood, S.M.; Gatter, K.C.; Harris, A.L.; Pugh, C.W.; et al. Induction of Endothelial PAS Domain Protein-1 by Hypoxia: Characterization and Comparison With Hypoxia-Inducible Factor-1a. Blood 1998, 92, 2260–2268. [Google Scholar] [CrossRef]
- Hartmann, H.; Eltzschig, H.K.; Wurz, H.; Hantke, K.; Rakin, A.; Yazdi, A.S.; Matteoli, G.; Bohn, E.; Autenrieth, I.B.; Karhausen, J.; et al. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 2008, 134, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Woo, K.J.; Lee, T.-J.; Park, J.-W.; Kwon, T.K. Desferrioxamine, an iron chelator, enhances HIF-1a accumulation via cyclooxygenase-2 signaling pathway. Biochem. Biophys. Res. Commun. 2006, 343, 8–14. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32. [Google Scholar] [CrossRef]
- Staples, K.J.; Sotoodehnejadnematalahi, F.; Pearson, H.; Frankenbergerb, M.; Francescut, L.; Ziegler-Heitbrock, L.; Burke, B. Monocyte-derived macrophages matured under prolonged hypoxia transcriptionally up-regulate HIF-1 mRNA. Immunobiology 2011, 216, 832–839. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhof, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Lua, J.; Tanb, M.; Caia, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007, 39, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting Edge: Essential Role of Hypoxia Inducible Factor-1 a in Development of Lipopolysaccharide-Induced Sepsis. J. Immunol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Melillo, G.; Taylor, L.S.; Brooks, A.; Musso, T.; Cox, G.W.; Varesio, L. Functional Requirement of the Hypoxia-responsive Element in the Activation of the Inducible Nitric Oxide Synthase Promoter by the Iron Chelator Desferrioxamine. J. Biol. Chem. 1997, 272, 12236–12243. [Google Scholar] [CrossRef] [Green Version]
- Nairz, M.; Weiss, G. Iron in infection and immunity. Mol. Aspects Med. 2020, 75, 1–18. [Google Scholar] [CrossRef]
- Baay-Guzman, G.J.; Duran-Padilla, M.A.; Rangel-Santiago, J.; Tirado-Rodriguez, B.; Antonio-Andres, G.; Barrios-Payan, J.; Mata-Espinosa, D.; Klunder-Klunder, M.; Vega, M.I.; Hernandez-Pando, R.; et al. Dual role of hypoxia-inducible factor 1 α in experimental pulmonary tuberculosis: Its implication as a new therapeutic target. Future Microbiol. 2018, 13, 785–798. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef] [Green Version]
- Braverman, J.; Sogi, K.M.; Benjamin, D.; Nomura, D.K.; Stanley, S.A. HIF-1alpha Is an Essential Mediator of IFN-gamma-Dependent Immunity to Mycobacterium tuberculosis. J. Immunol. 2016, 197, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1a–dependent NF-kB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Goldrath, A.W.; Bergh, J.; Johnson, R.S. An HIF-1a/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.L.; Koeller, D.M.; Ramin, V.C.; Klausner, R.D.; Harford, J.B. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3' untranslated region of the mRNA. EMBO J. 1989, 8, 3693–3699. [Google Scholar] [CrossRef]
- Theil, E.C. Iron regulatory elements (IREs): A family of mRNA non-coding sequences. Biochem. J. 1994, 304, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12. [Google Scholar] [CrossRef]
- Neves, J.; Haider, T.; Gassmann, M.; Muckenthaler, M.U. Iron Homeostasis in the Lungs—A Balance between Health and Disease. Pharmaceuticals 2019, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, T.; Hentze, M.W. Finding the hairpin in the haystack: Searching for RNA motifs. Treds Genet. 1995, 11. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; LaVaute, T.; Iwai, K.; Klausner, R.D.; Rouault, T.A. Identification of a conserved and functional iron-responsive element in the 5'-untranslated region of mammalian mitochondrial aconitase. J. Biol. Chem. 1996, 271, 24226–24230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalinske, K.L.; Chen, O.S.; Eisenstein, R.S. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J. Biol. Chem. 1998, 273, 3740–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheluccia, A.; Cordesa, T.; Ghelfia, J.; Pailota, A.; Reilingb, N.; Goldmannc, O.; Binza, T.; Wegnera, A.; Tallama, A.; Rausella, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Nat. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.; Chen, T.D.; Buang, N.; Olona, A.; Ko, J.H.; Prendecki, M.; Costa, A.S.H.; Nikitopoulou, E.; Tronci, L.; Pusey, C.D.; et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep. 2019, 28, 498–511, e495. [Google Scholar] [CrossRef] [Green Version]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556. [Google Scholar] [CrossRef]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Invest. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- DeBerge, M.; Lantz, C.; Dehn, S.; Sullivan, D.P.; Van der Laan, A.M.; Niessen, H.W.M.; Flanagan, M.E.; Brat, D.J.; Feinstein, M.J.; Kaushal, S.; et al. Hypoxia-inducible factors individually facilitate inflammatory myeloid metabolism and inefficient cardiac repair. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Elks, P.M.; Brizee, S.; van der Vaart, M.; Walmsley, S.R.; Van Eeden, F.J.; Renshaw, S.A.; Meijer, A.H. Hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PLoS Pathog. 2013, 9, e1003789. [Google Scholar] [CrossRef]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-mediated innate immune responses: Cell signaling and therapeutic implications. Hypoxia 2014, 2, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Evelyn, L.K.; Padberg, C.; Nora Koll, V.S.; Fandrey, J.; Winning, S. The Importance of Hypoxia-Inducible Factors (HIF-1 and HIF-2) for the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 1–26. [Google Scholar]
- Hsu, T.S.; Lin, Y.L.; Wang, Y.A.; Mo, S.T.; Chi, P.Y.; Lai, A.C.; Pan, H.Y.; Chang, Y.J.; Lai, M.Z. HIF-2alpha is indispensable for regulatory T cell function. Nat. Commun. 2020, 11, 5005. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Collins, H.L.; Kaufmann, S.H.E.; Schaible, U.E. Iron Chelation Via Deferoxamine Exacerbates Experimental Salmonellosis Via Inhibition of the Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Dependent Respiratory Burst. J. Immunol. 2002, 3458–3463. [Google Scholar] [CrossRef]
- Han, Z.; Varadharaj, S.; Giedt, R.J.; Zweier, J.L.; Szeto, H.H.; Alevriadou, B.R. Mitochondria-Derived Reactive Oxygen Species Mediate Heme Oxygenase-1 Expression in Sheared Endothelial Cells. J. Pharmacol. Exp. Ther. 2009, 329, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Calzi, S.L.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell–derived factor 1 promotes angiogenesis via a heme oxygenase 1–dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef]
- Bussolati, B.; Ahmed, A.; Pemberton, H.; Landis, R.C.; Carlo, F.D.; Haskard, D.O.; Mason, J.C. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: Induction of angiogenesis and inhibition of leukocytic infiltration. Blood 2004, 103, 761–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Guan, S.-S.; Yang, H.-J.; Chang, C.-C.; Luo, T.-Y.; Chang, J.; Ho, A.-S. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxiamediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J. Biomed. Sci. 2016, 23, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Fuyuhiko, M.; Yamauchi, J.-I.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef]
- Shang, F.-t.; Hui, L.-l.; An, X.-s.; Zhang, X.-c.; Guo, S.-g.; Kui, Z. ZnPPIX inhibits peritoneal metastasis of gastric cancer via its antiangiogenic activity. Biomed. Pharmacother. 2015, 71, 240–246. [Google Scholar] [CrossRef]
- Nemeth, Z.; Li, M.; Csizmadia, E.; Döme, B.; Johansson, M.; Persson, J.L.; Seth, P.; Otterbein, L.; Wegiel, B. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget 2015, 6, 33675–33688. [Google Scholar] [CrossRef] [Green Version]
- Hoang, K.N.L.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-C.; Chiang, S.-K.; Chen, S.-E.; Yu, Y.-L.; Chou, R.-H.; Chang, W.-C. Heme oxygenase-1 mediates BAY 11e7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, A.S.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization and function in health and disease. J. Cell. Physiol. 2018, 223, 6425–6440. [Google Scholar] [CrossRef]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [Green Version]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; Pizzol, M.D.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Zhou, Y.; Que, K.-T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.-P.; Liu, Z.-J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [Green Version]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [Green Version]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [Green Version]
- Nairz, M.; Fritsche, G.; Brunner, P.; Talasz, H.; Hantke, K.; Weiss, G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 2008, 38, 1923–1936. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Ludwiczek, S.; Theurl, M.; Mair, S.M.; Fritsche, G.; Weiss, G. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell. Microbiol. 2007, 9, 2126–2140. [Google Scholar] [CrossRef]
- Weiss, G.; Werner-Felmayer, G.; Werner, E.R.; Grunewald, K.; Wachter, H.; Hentze, M.W. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 1994, 180, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittala, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Pantopoulos, K. Hepcidin Therapeutics. Pharmaceuticals (Basel) 2018, 11, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 2016, 7, 160. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, J.; Denadai, M.B.; Costa, D.L. Crosstalk between Heme Oxygenase-1 and Iron Metabolism in Macrophages: Implications for the Modulation of Inflammation and Immunity. Antioxidants 2022, 11, 861. https://doi.org/10.3390/antiox11050861
de Oliveira J, Denadai MB, Costa DL. Crosstalk between Heme Oxygenase-1 and Iron Metabolism in Macrophages: Implications for the Modulation of Inflammation and Immunity. Antioxidants. 2022; 11(5):861. https://doi.org/10.3390/antiox11050861
Chicago/Turabian Stylede Oliveira, Joseana, Marina B. Denadai, and Diego L. Costa. 2022. "Crosstalk between Heme Oxygenase-1 and Iron Metabolism in Macrophages: Implications for the Modulation of Inflammation and Immunity" Antioxidants 11, no. 5: 861. https://doi.org/10.3390/antiox11050861
APA Stylede Oliveira, J., Denadai, M. B., & Costa, D. L. (2022). Crosstalk between Heme Oxygenase-1 and Iron Metabolism in Macrophages: Implications for the Modulation of Inflammation and Immunity. Antioxidants, 11(5), 861. https://doi.org/10.3390/antiox11050861