
New Progress in the Molecular Regulations and Therapeutic Applications in Cardiac Oxidative Damage Caused by Pressure Overload



Abstract
:1. Introduction
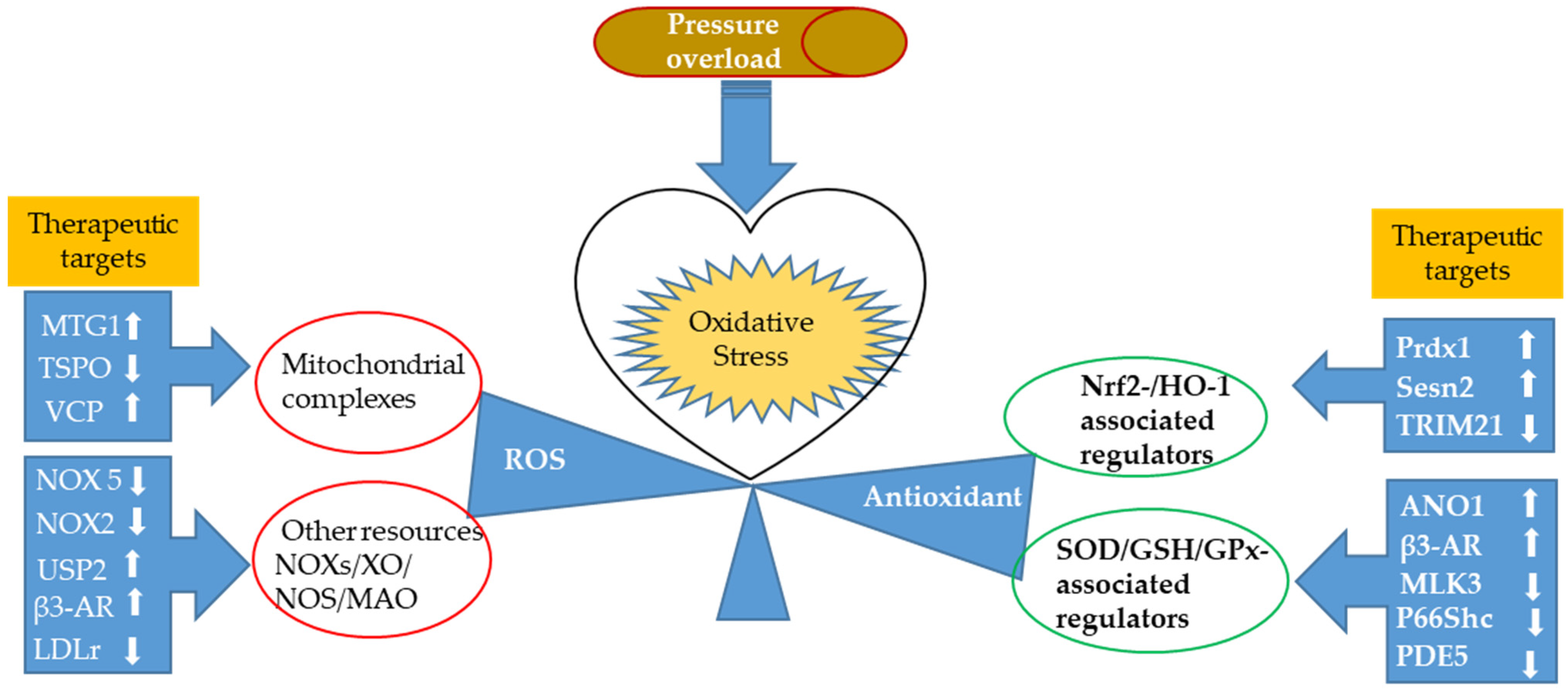
2. Oxidative Stress in Response to Cardiac Pressure Overload
2.1. ROS Production in Experimental Models of Pressure Overload
2.1.1. Newly Identified Proteins Associated with Mitochondrial Ros Production and Related Regulating Mechanisms
2.1.2. Nox-Associated ROS Production in a Pressure-Overloaded Heart
2.2. Antioxidant Response in Experimental Models of Pressure Overload
2.2.1. Nuclear Factor-Erythroid Factor 2-Related Factor 2 (Nrf2)-Associated Regulators
2.2.2. Newly Discovered Regulators of the Antioxidant Defense System in the Heart Primarily through Modulating First-Line Defense Antioxidants
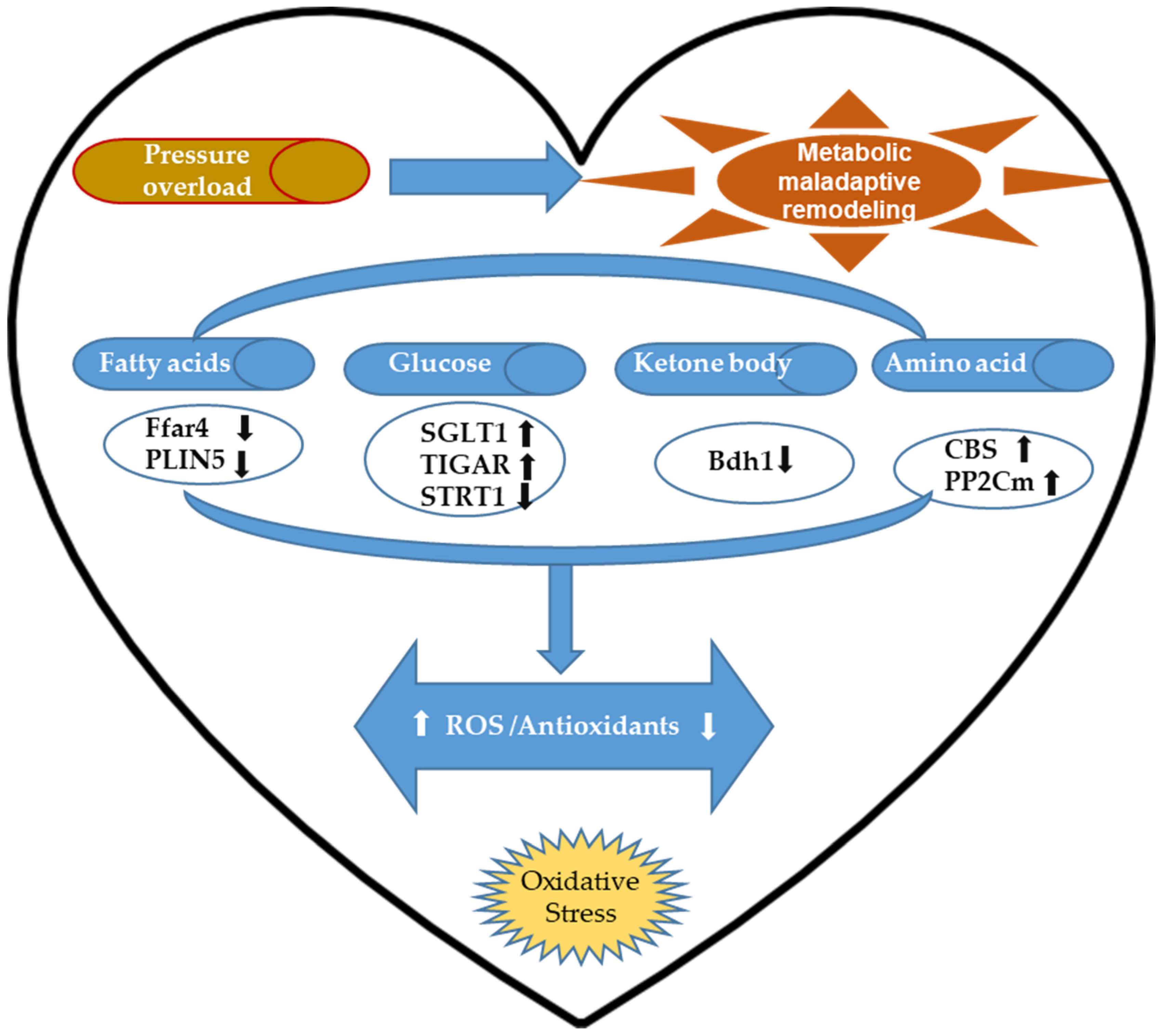
2.3. Cardiac Metabolic Remodeling-Associated Oxidative Stress and Antioxidative Regulation upon Pressure Overload
3. Newly Developed Antioxidative Therapeutic Approaches against Cardiac Pressure Overload
3.1. Repurposed Pharmacological Agents
3.2. Naturally Derived Organic Extracts
3.3. Natural Organic Compounds
3.4. Potential Chemical Compounds with Antioxidant Property
3.5. Calorie Restriction
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxidative Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Atika, E.; Naouel, E. Endogenous Enzymatic Antioxidant Defense and Pathologies. In Antioxidants; Waisundara, V., Ed.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Pitoulis, F.G.; Terracciano, C.M. Heart Plasticity in Response to Pressure- and Volume-Overload: A Review of Findings in Compensated and Decompensated Phenotypes. Front. Physiol. 2020, 11, 92. [Google Scholar] [CrossRef]
- Nabben, M.; Luiken, J.; Glatz, J.F.C. Metabolic remodelling in heart failure revisited. Nat. Rev. Cardiol. 2018, 15, 780. [Google Scholar] [CrossRef]
- Matsushita, N.; Ishida, N.; Ibi, M.; Saito, M.; Sanbe, A.; Shimojo, H.; Suzuki, S.; Koepsell, H.; Takeishi, Y.; Morino, Y.; et al. Chronic Pressure Overload Induces Cardiac Hypertrophy and Fibrosis via Increases in SGLT1 and IL-18 Gene Expression in Mice. Int. Heart J. 2018, 59, 1123–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, M.; Baicu, C.F.; Van Laer, A.O.; Zhang, Y.; McDonald, L.T.; LaRue, A.C.; Zile, M.R.; Bradshaw, A.D. Pressure overload generates a cardiac-specific profile of inflammatory mediators. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H331–H340. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Moskalik, A.; Niderla-Bielińska, J.; Ratajska, A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 2021, 1–18. [Google Scholar] [CrossRef]
- Chi, R.F.; Li, L.; Wang, A.L.; Yang, H.; Xi, J.; Zhu, Z.F.; Wang, K.; Li, B.; Yang, L.G.; Qin, F.Z.; et al. Enhanced oxidative stress mediates pathological autophagy and necroptosis in cardiac myocytes in pressure overload induced heart failure in rats. Clin. Exp. Pharmacol. Physiol. 2022, 49, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, A.C.; Sag, C.M.; Maier, L.S. Reactive oxygen species and excitation-contraction coupling in the context of cardiac pathology. J. Mol. Cell. Cardiol. 2014, 73, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Rodrigo, R.; Prieto, J.C.; Aguayo, R.; Ramos, C.; Puentes, Á.; Gajardo, A.; Panieri, E.; Rojas-Solé, C.; Lillo-Moya, J.; Saso, L. Joint Cardioprotective Effect of Vitamin C and Other Antioxidants against Reperfusion Injury in Patients with Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Molecules 2021, 26, 5702. [Google Scholar] [CrossRef]
- Li, B.; Sun, Y.; Wang, J.P.; Chi, R.F.; Wang, K.; Yang, Z.J.; Qin, F.Z.; Fan, B. Antioxidant N-acetylcysteine inhibits maladaptive myocyte autophagy in pressure overload induced cardiac remodeling in rats. Eur. J. Pharmacol. 2018, 839, 47–56. [Google Scholar] [CrossRef]
- Saheera, S.; Potnuri, A.G.; Nair, R.R. Protective effect of antioxidant Tempol on cardiac stem cells in chronic pressure overload hypertrophy. Life Sci. 2019, 222, 88–93. [Google Scholar] [CrossRef]
- Kumar, V.; A., A.K.; Sanawar, R.; Jaleel, A.; Santhosh Kumar, T.R.; Kartha, C.C. Chronic Pressure Overload Results in Deficiency of Mitochondrial Membrane Transporter ABCB7 Which Contributes to Iron Overload, Mitochondrial Dysfunction, Metabolic Shift and Worsens Cardiac Function. Sci. Rep. 2019, 9, 13170. [Google Scholar] [CrossRef] [Green Version]
- Geraets, I.M.E.; Glatz, J.F.C.; Luiken, J.; Nabben, M. Pivotal role of membrane substrate transporters on the metabolic alterations in the pressure-overloaded heart. Cardiovasc. Res. 2019, 115, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhao, Y.; Weng, X.; Lu, Y.; Li, W.; Tang, K.; Chen, W.; Liu, Z.; Qi, X.; Zheng, J.; et al. Novel role of mitochondrial GTPases 1 in pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2019, 128, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Thai, P.N.; Daugherty, D.J.; Frederich, B.J.; Lu, X.; Deng, W.; Bers, D.M.; Dedkova, E.N.; Schaefer, S. Cardiac-specific Conditional Knockout of the 18-kDa Mitochondrial Translocator Protein Protects from Pressure Overload Induced Heart Failure. Sci. Rep. 2018, 8, 16213. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Genomic characterization reveals novel mechanisms underlying the valosin-containing protein-mediated cardiac protection against heart failure. Redox Biol. 2020, 36, 101662. [Google Scholar] [CrossRef] [PubMed]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Anagnostopoulou, A.; Camargo, L.L.; Rios, F.J.; Montezano, A.C. Vascular Biology of Superoxide-Generating NADPH Oxidase 5-Implications in Hypertension and Cardiovascular Disease. Antioxid. Redox Signal. 2019, 30, 1027–1040. [Google Scholar] [CrossRef]
- Montezano, A.C.; De Lucca Camargo, L.; Persson, P.; Rios, F.J.; Harvey, A.P.; Anagnostopoulou, A.; Palacios, R.; Gandara, A.C.P.; Alves-Lopes, R.; Neves, K.B.; et al. NADPH Oxidase 5 Is a Pro-Contractile Nox Isoform and a Point of Cross-Talk for Calcium and Redox Signaling-Implications in Vascular Function. J. Am. Heart Assoc. 2018, 7, e009388. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.J.; Zhao, C.L.; Ouyang, S.; Deng, K.Q.; Zhu, L.; Montezano, A.C.; Zhang, C.; Hu, F.; Zhu, X.Y.; Tian, S.; et al. Ca(2+)-Dependent NOX5 (NADPH Oxidase 5) Exaggerates Cardiac Hypertrophy Through Reactive Oxygen Species Production. Hypertension 2020, 76, 827–838. [Google Scholar] [CrossRef]
- Parajuli, N.; Patel, V.B.; Wang, W.; Basu, R.; Oudit, G.Y. Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin. Sci. 2014, 127, 331–340. [Google Scholar] [CrossRef]
- Xing, J.; Li, P.; Hong, J.; Wang, M.; Liu, Y.; Gao, Y.; Dong, J.; Gu, H.; Li, L. Overexpression of Ubiquitin-Specific Protease 2 (USP2) in the Heart Suppressed Pressure Overload-Induced Cardiac Remodeling. Mediat. Inflamm. 2020, 2020, 4121750. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Chen, J.; Qin, C.; Liu, J.; Guo, D.; Wang, R.; Hu, J.; Zou, Q.; Yang, J.; et al. Beta3-Adrenergic Receptor Activation Alleviates Cardiac Dysfunction in Cardiac Hypertrophy by Regulating Oxidative Stress. Oxidative Med. Cell. Longev. 2021, 2021, 3417242. [Google Scholar] [CrossRef] [PubMed]
- Muthuramu, I.; Mishra, M.; Aboumsallem, J.P.; Postnov, A.; Gheysens, O.; De Geest, B. Cholesterol lowering attenuates pressure overload-induced heart failure in mice with mild hypercholesterolemia. Aging 2019, 11, 6872–6891. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Smyrnias, I.; Zhang, X.; Zhang, M.; Murray, T.V.; Brandes, R.P.; Schröder, K.; Brewer, A.C.; Shah, A.M. Nicotinamide adenine dinucleotide phosphate oxidase-4-dependent upregulation of nuclear factor erythroid-derived 2-like 2 protects the heart during chronic pressure overload. Hypertension 2015, 65, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Yin, G.; Huang, C.; Wang, H.; Gao, J.; Luo, J.; Zhang, Z.; Wang, J.; Hong, J.; Chai, X. Peroxiredoxin-1 ameliorates pressure overload-induced cardiac hypertrophy and fibrosis. Biomed. Pharmacother. 2020, 129, 110357. [Google Scholar] [CrossRef]
- Zhang, N.; Liao, H.H.; Feng, H.; Mou, S.Q.; Li, W.J.; Aiyasiding, X.; Lin, Z.; Ding, W.; Zhou, Z.Y.; Yan, H.; et al. Knockout of AMPKα2 Blocked the Protection of Sestrin2 Overexpression Against Cardiac Hypertrophy Induced by Pressure Overload. Front. Pharmacol. 2021, 12, 716884. [Google Scholar] [CrossRef]
- Pan, J.A.; Sun, Y.; Jiang, Y.P.; Bott, A.J.; Jaber, N.; Dou, Z.; Yang, B.; Chen, J.S.; Catanzaro, J.M.; Du, C.; et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol. Cell 2016, 61, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.C.; Miao, W.Q.; Wang, Y.; Zhou, S.F. ANO1 relieves pressure overload-induced myocardial fibrosis in mice by inhibiting TGF-β/Smad3 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8493–8501. [Google Scholar] [CrossRef]
- He, S.; Liu, S.; Wu, X.; Xin, M.; Ding, S.; Xin, D.; Ouyang, H.; Zhang, J. Protective role of downregulated MLK3 in myocardial adaptation to chronic hypoxia. J. Physiol. Biochem. 2016, 73, 371–380. [Google Scholar] [CrossRef]
- Calamaras, T.D.; Baumgartner, R.A.; Aronovitz, M.J.; McLaughlin, A.L.; Tam, K.; Richards, D.A.; Cooper, C.W.; Li, N.; Baur, W.E.; Qiao, X.; et al. Mixed lineage kinase-3 prevents cardiac dysfunction and structural remodeling with pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H145–H159. [Google Scholar] [CrossRef]
- Wang, J.; Deng, B.; Liu, Q.; Huang, Y.; Chen, W.; Li, J.; Zhou, Z.; Zhang, L.; Liang, B.; He, J.; et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020, 11, 574. [Google Scholar] [CrossRef]
- Wang, S.Y.; Zhu, S.; Wu, J.; Zhang, M.; Xu, Y.; Xu, W.; Cui, J.; Yu, B.; Cao, W.; Liu, J. Exercise enhances cardiac function by improving mitochondrial dysfunction and maintaining energy homoeostasis in the development of diabetic cardiomyopathy. J. Mol. Med. 2020, 98, 245–261. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, H.; Liu, J. P66Shc Deletion Ameliorates Oxidative Stress and Cardiac Dysfunction in Pressure Overload-Induced Heart Failure. J. Card. Fail. 2020, 26, 243–253. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.Q.; Liu, F.Y.; Guo, Z.; An, P.; Wang, M.Y.; Yang, Z.; Fan, D.; Tang, Q.Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2021, 8, 822969. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Murphy, K.A.; Harsch, B.A.; Healy, C.L.; Joshi, S.S.; Huang, S.; Walker, R.E.; Wagner, B.M.; Ernste, K.M.; Huang, W.; Block, R.C.; et al. Free fatty acid receptor 4 responds to endogenous fatty acids to protect the heart from pressure overload. Cardiovasc. Res. 2021, 118, 1061–1073. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, Y.; Wu, J.; Zhao, Y.; Gao, X.; Chen, Y.; Sun, C.; Xiao, L.; Zheng, P.; Hu, P.; et al. Plin5 deficiency exacerbates pressure overload-induced cardiac hypertrophy and heart failure by enhancing myocardial fatty acid oxidation and oxidative stress. Free Radic. Biol. Med. 2019, 141, 372–382. [Google Scholar] [CrossRef]
- Sayour, A.A.; Ruppert, M.; Oláh, A.; Benke, K.; Barta, B.A.; Zsáry, E.; Ke, H.; Horváth, E.M.; Merkely, B.; Radovits, T. Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure. Antioxidants 2021, 10, 1190. [Google Scholar] [CrossRef]
- Okawa, Y.; Hoshino, A.; Ariyoshi, M.; Kaimoto, S.; Tateishi, S.; Ono, K.; Uchihashi, M.; Iwai-Kanai, E.; Matoba, S. Ablation of cardiac TIGAR preserves myocardial energetics and cardiac function in the pressure overload heart failure model. Am. J. Physiol. Circ. Physiol. 2019, 316, H1366–H1377. [Google Scholar] [CrossRef]
- Sanz, M.N.; Grimbert, L.; Moulin, M.; Gressette, M.; Rucker-Martin, C.; Lemaire, C.; Mericskay, M.; Veksler, V.; Ventura-Clapier, R.; Garnier, A.; et al. Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. Int. J. Mol. Sci. 2019, 20, 5005. [Google Scholar] [CrossRef] [Green Version]
- Uchihashi, M.; Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Tateishi, S.; Ono, K.; Yamanaka, R.; Hato, D.; Fushimura, Y.; et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload-Induced Heart Failure. Circulation Heart Fail. 2017, 10, e004417. [Google Scholar] [CrossRef]
- Liu, J.; Quan, J.; Li, Y.; Wu, Y.; Yang, L. Blood homocysteine levels could predict major adverse cardiac events in patients with acute coronary syndrome: A STROBE-compliant observational study. Medicine 2018, 97, e12626. [Google Scholar] [CrossRef]
- Muthuramu, I.; Singh, N.; Amin, R.; Nefyodova, E.; Debasse, M.; Van Horenbeeck, I.; Jacobs, F.; De Geest, B. Selective homocysteine-lowering gene transfer attenuates pressure overload-induced cardiomyopathy via reduced oxidative stress. J. Mol. Med. 2015, 93, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Huo, S.; Shi, W.; Ma, H.; Yan, D.; Luo, P.; Guo, J.; Li, C.; Lin, J.; Zhang, C.; Li, S.; et al. Alleviation of Inflammation and Oxidative Stress in Pressure Overload-Induced Cardiac Remodeling and Heart Failure via IL-6/STAT3 Inhibition by Raloxifene. Oxidative Med. Cell. Longev. 2021, 2021, 6699054. [Google Scholar] [CrossRef]
- Guan, P.; Liang, Y.; Wang, N. Fasudil alleviates pressure overload-induced heart failure by activating Nrf2-mediated antioxidant responses. J. Cell. Biochem. 2018, 119, 6452–6460. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F.; Zhang, Y.; Kang, Y.; Wang, H.; Si, M.; Su, L.; Xin, X.; Xue, F.; Hao, F.; et al. Celecoxib prevents pressure overload-induced cardiac hypertrophy and dysfunction by inhibiting inflammation, apoptosis and oxidative stress. J. Cell. Mol. Med. 2016, 20, 116–127. [Google Scholar] [CrossRef]
- Peng, S.; Lu, X.F.; Qi, Y.D.; Li, J.; Xu, J.; Yuan, T.Y.; Wu, X.Y.; Ding, Y.; Li, W.H.; Zhou, G.Q.; et al. LCZ696 Ameliorates Oxidative Stress and Pressure Overload-Induced Pathological Cardiac Remodeling by Regulating the Sirt3/MnSOD Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 9815039. [Google Scholar] [CrossRef]
- Li, X.; Braza, J.; Mende, U.; Choudhary, G.; Zhang, P. Cardioprotective effects of early intervention with sacubitril/valsartan on pressure overloaded rat hearts. Sci. Rep. 2021, 11, 16542. [Google Scholar] [CrossRef]
- Nicolas, D.; Kerndt, C.C.; Reed, M. Sacubitril/Valsartan. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Xu, M.; Wan, C.X.; Huang, S.H.; Wang, H.B.; Fan, D.; Wu, H.M.; Wu, Q.Q.; Ma, Z.G.; Deng, W.; Tang, Q.Z. Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy. Cell Death Dis. 2019, 10, 403. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Lu, Y.; Ping, N.N.; Li, X.; Lin, Y.X.; Li, C.F. Apocynin ameliorates pressure overload-induced cardiac remodeling by inhibiting oxidative stress and apoptosis. Physiol. Res. 2017, 66, 741–752. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, J.; An, W.; Lin, Y.; Yang, Y.; Zang, W. Apocynin attenuates pressure overload-induced cardiac hypertrophy in rats by reducing levels of reactive oxygen species. Can. J. Physiol. Pharmacol. 2010, 88, 745–752. [Google Scholar] [CrossRef]
- Wei, Y.J.; Xu, H.J.; Chen, J.J.; Yang, X.; Xiong, J.; Wang, J.; Cheng, F. Carnosic acid protects against pressure overload-induced cardiac remodelling by inhibiting the AKT/GSK3β/NOX4 signalling pathway. Exp. Ther. Med. 2020, 20, 3709–3719. [Google Scholar] [CrossRef]
- Cao, T.T.; Chen, H.H.; Dong, Z.; Xu, Y.W.; Zhao, P.; Guo, W.; Wei, H.C.; Zhang, C.; Lu, R. Stachydrine Protects Against Pressure Overload-Induced Cardiac Hypertrophy by Suppressing Autophagy. Cell. Physiol. Biochem. 2017, 42, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wei, W.Y.; Yang, Z.; Che, Y.; Jin, Y.G.; Liao, H.H.; Wang, S.S.; Deng, W.; Tang, Q.Z. Nobiletin, a Polymethoxy Flavonoid, Protects Against Cardiac Hypertrophy Induced by Pressure-Overload via Inhibition of NAPDH Oxidases and Endoplasmic Reticulum Stress. Cell. Physiol. Biochem. 2017, 42, 1313–1325. [Google Scholar] [CrossRef]
- Liao, L.Z.; Chen, Z.C.; Wang, S.S.; Liu, W.B.; Zhao, C.L.; Zhuang, X.D. NLRP3 inflammasome activation contributes to the pathogenesis of cardiocytes aging. Aging 2021, 13, 20534–20551. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.; Li, M.; Wang, Z.; Li, B.; Qu, X.; Chen, S. Cardamonin Alleviates Pressure Overload-induced Cardiac Remodeling and Dysfunction Through Inhibition of Oxidative Stress. J. Cardiovasc. Pharmacol. 2016, 68, 441–451. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Xiao, Y.; Duan, M.X.; Yuan, Y.; Jiang, X.H.; Yang, Z.; Liao, H.H.; Deng, W.; Tang, Q.Z. Aucubin protects against pressure overload-induced cardiac remodelling via the β(3) -adrenoceptor-neuronal NOS cascades. Br. J. Pharmacol. 2018, 175, 1548–1566. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xie, Z.; Jiang, N.; Wu, Z.; Xue, R.; Dong, B.; Fan, W.; Dai, G.; Chen, C.; Li, J.; et al. Hispidulin Attenuates Cardiac Hypertrophy by Improving Mitochondrial Dysfunction. Front. Cardiovasc. Med. 2020, 7, 582890. [Google Scholar] [CrossRef]
- Pop, C.; Berce, C.; Ghibu, S.; Scurtu, I.; Sorițău, O.; Login, C.; Kiss, B.; Ștefan, M.G.; Fizeșan, I.; Silaghi, H.; et al. Effects of Lycium barbarum L. Polysaccharides on Inflammation and Oxidative Stress Markers in a Pressure Overload-Induced Heart Failure Rat Model. Molecules 2020, 25, 466. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Zhao, J.; Dong, B.; Cai, X.; Jiang, J.; Xue, R.; Yao, F.; Dong, Y.; Liu, C. Lycopene protects against pressure overload-induced cardiac hypertrophy by attenuating oxidative stress. J. Nutr. Biochem. 2019, 66, 70–78. [Google Scholar] [CrossRef]
- Dong, B.; Liu, C.; Xue, R.; Wang, Y.; Sun, Y.; Liang, Z.; Fan, W.; Jiang, J.; Zhao, J.; Su, Q.; et al. Fisetin inhibits cardiac hypertrophy by suppressing oxidative stress. J. Nutr. Biochem. 2018, 62, 221–229. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, X.; Zhang, Y.L.; Bai, J.; Hidru, T.H.; Wang, Q.S.; Li, H.H. Vitamin D attenuates pressure overload-induced cardiac remodeling and dysfunction in mice. J. Steroid Biochem. Mol. Biol. 2018, 178, 293–302. [Google Scholar] [CrossRef]
- Peng, Q.; Ding, R.; Wang, X.; Yang, P.; Jiang, F.; Chen, X. Effect of Irisin on Pressure Overload-Induced Cardiac Remodeling. Arch. Med. Res. 2021, 52, 182–190. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Zhao, X.; Xiao, L.; Yi, S.; Kong, Y.; Jiang, Y.; Zhang, J. A cathelicidin-related antimicrobial peptide suppresses cardiac hypertrophy induced by pressure overload by regulating IGFR1/PI3K/AKT and TLR9/AMPKα. Cell Death Dis. 2020, 11, 96. [Google Scholar] [CrossRef]
- Bai, W.; Ren, M.; Cheng, W.; Lu, X.; Liu, D.; Wang, B. Qindan Capsule Attenuates Myocardial Hypertrophy and Fibrosis in Pressure Overload-Induced Mice Involving mTOR and TGF-β1/Smad Signaling Pathway Inhibition. Evid-Based Complementary Altern. Med. 2021, 2021, 5577875. [Google Scholar] [CrossRef]
- Liu, J.; Ai, Y.; Niu, X.; Shang, F.; Li, Z.; Liu, H.; Li, W.; Ma, W.; Chen, R.; Wei, T.; et al. Taurine protects against cardiac dysfunction induced by pressure overload through SIRT1-p53 activation. Chem. Biol. Interact. 2020, 317, 108972. [Google Scholar] [CrossRef]
- Kumar, A.; Supowit, S.; Potts, J.D.; DiPette, D.J. Alpha-calcitonin gene-related peptide prevents pressure-overload induced heart failure: Role of apoptosis and oxidative stress. Physiol. Rep. 2019, 7, e14269. [Google Scholar] [CrossRef]
- Triastuti, E.; Nugroho, A.B.; Zi, M.; Prehar, S.; Kohar, Y.S.; Bui, T.A.; Stafford, N.; Cartwright, E.J.; Abraham, S.; Oceandy, D. Pharmacological inhibition of Hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br. J. Pharmacol. 2019, 176, 3956–3971. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, H.; Li, Y.; Tian, J.; Deng, S. Wnt-C59 Attenuates Pressure Overload-Induced Cardiac Hypertrophy via Interruption of Wnt Pathway. Med. Sci. Monit. 2020, 26, e923025. [Google Scholar] [CrossRef]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef]
- Ngwenyama, N.; Kirabo, A.; Aronovitz, M.; Velázquez, F.; Carrillo-Salinas, F.; Salvador, A.M.; Nevers, T.; Amarnath, V.; Tai, A.; Blanton, R.M.; et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation 2021, 143, 1242–1255. [Google Scholar] [CrossRef]
- Shang, L.; Weng, X.; Wang, D.; Yue, W.; Mernaugh, R.; Amarnath, V.; Weir, E.K.; Dudley, S.C.; Xu, Y.; Hou, M.; et al. Isolevuglandin scavenger attenuates pressure overload-induced cardiac oxidative stress, cardiac hypertrophy, heart failure and lung remodeling. Free Radic. Biol. Med. 2019, 141, 291–298. [Google Scholar] [CrossRef]
- Kobara, M.; Furumori-Yukiya, A.; Kitamura, M.; Matsumura, M.; Ohigashi, M.; Toba, H.; Nakata, T. Short-Term Caloric Restriction Suppresses Cardiac Oxidative Stress and Hypertrophy Caused by Chronic Pressure Overload. J. Card. Fail. 2015, 21, 656–666. [Google Scholar] [CrossRef]
- Kobara, M.; Naseratun, N.; Toba, H.; Nakata, T. Preconditioning with Short-term Dietary Restriction Attenuates Cardiac Oxidative Stress and Hypertrophy Induced by Chronic Pressure Overload. Nutrients 2021, 13, 737. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The quest for selective nox inhibitors and therapeutics: Challenges, triumphs and pitfalls. Antioxid. Redox Signal. 2014, 20, 2741–2754. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.M.; Ram, C.; Murty, U.S.; Sahu, B.D. A review on herbal Nrf2 activators with preclinical evidence in cardiovascular diseases. Phytother. Res. 2021, 35, 5068–5102. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Cong, S.; Chan, X.; Yap, E.P.; Yu, F.; Hausenloy, D.J. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312. [Google Scholar] [CrossRef]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Rabinovitch, P.S. Chapter 15—Autophagy and Proteostasis in Cardiac Aging. In Autophagy and Cardiometabolic Diseases; Ren, J., Sowers, J.R., Zhang, Y., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 171–186. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic Approaches | Route of Administration | Animal Model | Targeted Antioxidative Mechanisms in the Heart | References | |
|---|---|---|---|---|---|
| Repurposed pharmacological agents | Raloxifene | Oral gavage | TAC | ↑SOD expression and activity and ↓iNOS expression via IL-6/STAT3 signaling | [58] |
| Fasudil | Subcutaneous injection | TAC | ↑Nrf2/HO-1 pathway and↑SOD, CAT, and GPx activities | [59] | |
| Celecoxib | Oral gavage | AAC | ↑Nrf2-mediated HO-1, NQO-1, and ↓MDA | [60] | |
| Sacubitril/Valsartan (LCZ696) | Oral gavage | TAC | ↓Superoxide and peroxide derivatives and ↑MnSOD and Sirt3 | [61,62,63] | |
| Naturally derived organic extracts | Oridonin | Oral gavage | AB | ↓gp91phox, p67phox and ROS production; ↑HO-1, SOD, and GPx | [64] |
| Apocynin | Voluntary oral ingestion | AAC | ↓Nox activity, O2−, and MDA; ↑SOD activity | [65,66] | |
| Carnosic acid (CA) | Oral gavage | AB | ↓AKT/GSK3β/Nox4 signaling; ↑SOD activity | [67] | |
| Stachydrine | Oral gavage | TAC | ↓gp91phox and p67phox expression, p47phoxphosphorylation, and p47phox/gp91phox colocalization | [68] | |
| Nobiletin (NOB) | Oral gavage | AB | ↑SOD1 concentration; ↓Nox2 and Nox4 expression and 4-HNE levels | [69] | |
| Astragaloside IV (AS-IV) | Intraperitoneal injection | TAC | ↓H2O2 content | [70] | |
| Cardamonin (Cam) | Intraperitoneal injection | TAC | ↓4-HNE and MDA; ↑SOD and GSH content | [71] | |
| Aucubin (AUB) | Intraperitoneal injection | AB | ↓ROS generation, P67phox expression, and lipid peroxidation; ↑SOD, GPx, and nNOS expression | [72] | |
| Hispidulin | Intraperitoneal injection | AB | ↑SOD1, MnSOD, and CAT expression | [73] | |
| Natural organic compounds | L. barbarum L. polysaccharides (LBPs) | Oral gavage | AAB | ↓plasma MDA levels | [74] |
| Lycopene | Oral gavage | AB | ↑ARE activity and ARE-mediated HO-1, SOD1, and CAT expression; ↓ROS production | [75] | |
| Fisetin | Intraperitoneal injection | AB | ↓ROS production | [76] | |
| Vitamin D (VD) | Oral gavage | TAC | ↓superoxide production, Nox2, Nox4, and p22phox expression | [77] | |
| Irisin | Intravenous injection | TAC | ↓Nox2 and XO; ↑SOD1 and plasma GPx | [78] | |
| Cathelicidin-related antimicrobial peptide (CRAMP) | Intraperitoneal injection | AB | ↑SOD2 and GPx activity; ↓Nox2 and Nox4 expression | [79] | |
| Qindan capsule (QC) | Oral gavage | TAC | ↓8-OHdG, MDA, and 15-isoprostane F2t | [80] | |
| Taurine | Oral gavage | TAC | ↓ROS production and MDA expression; ↑SOD expression | [81] | |
| Potential chemical compounds with antioxidant property | Alpha-calcitonin gene-related peptide (a-CGRP) | Subcutaneous injection | TAC | ↓4-HNE, 8-OHdG, and MDA; ↑total GSH | [82] |
| XMU-MP-1 | Intraperitoneal injection | TAC | ↑enhanced cell survival against H2O2 | [83] | |
| Wnt-C59 | Oral gavage | TAC | ↓ROS production and lipid peroxidation; ↑GPx and SOD activity | [84] | |
| Mitoquinone (MitoQ) | Oral gavage | AAC | ↓MDA levels via redox-sensitive Plscr4-miR-214 axis | [85] | |
| 2-hydroxybenzylamine (2-HOBA) | Oral gavage | TAC | ↓ROS production | [86,87] | |
| Non-pharmacological interventions | Calorie restriction (CR) | Voluntary oral ingestion | AAC | ↓8-OHdG, mitochondrial content of lipid hydroperoxide, Nox-dependent and mitochondrial superoxide production; ↑GPx and SOD activities | [88] |
| Dietary restriction preconditioning (DRPC) | Voluntary oral ingestion | AAC | ↓8-OHdG, mitochondrial content of lipid hydroperoxide, Nox-dependent, and mitochondrial superoxide production | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.; Dorsey, A.; Qiu, H. New Progress in the Molecular Regulations and Therapeutic Applications in Cardiac Oxidative Damage Caused by Pressure Overload. Antioxidants 2022, 11, 877. https://doi.org/10.3390/antiox11050877
Shi X, Dorsey A, Qiu H. New Progress in the Molecular Regulations and Therapeutic Applications in Cardiac Oxidative Damage Caused by Pressure Overload. Antioxidants. 2022; 11(5):877. https://doi.org/10.3390/antiox11050877
Chicago/Turabian StyleShi, Xiaomeng, Arin Dorsey, and Hongyu Qiu. 2022. "New Progress in the Molecular Regulations and Therapeutic Applications in Cardiac Oxidative Damage Caused by Pressure Overload" Antioxidants 11, no. 5: 877. https://doi.org/10.3390/antiox11050877
APA StyleShi, X., Dorsey, A., & Qiu, H. (2022). New Progress in the Molecular Regulations and Therapeutic Applications in Cardiac Oxidative Damage Caused by Pressure Overload. Antioxidants, 11(5), 877. https://doi.org/10.3390/antiox11050877