
Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations

 , , , , ,
, , , , ,  ,
, 
Abstract
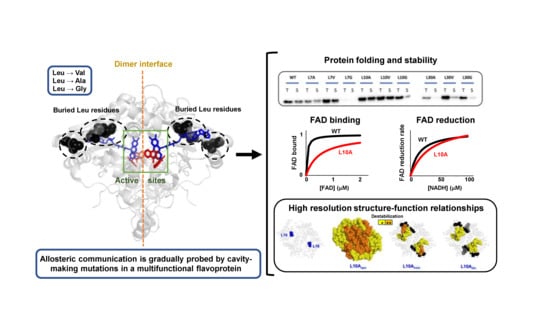
:
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Thermal Stability
2.3. Partial Proteolysis by Thermolysin
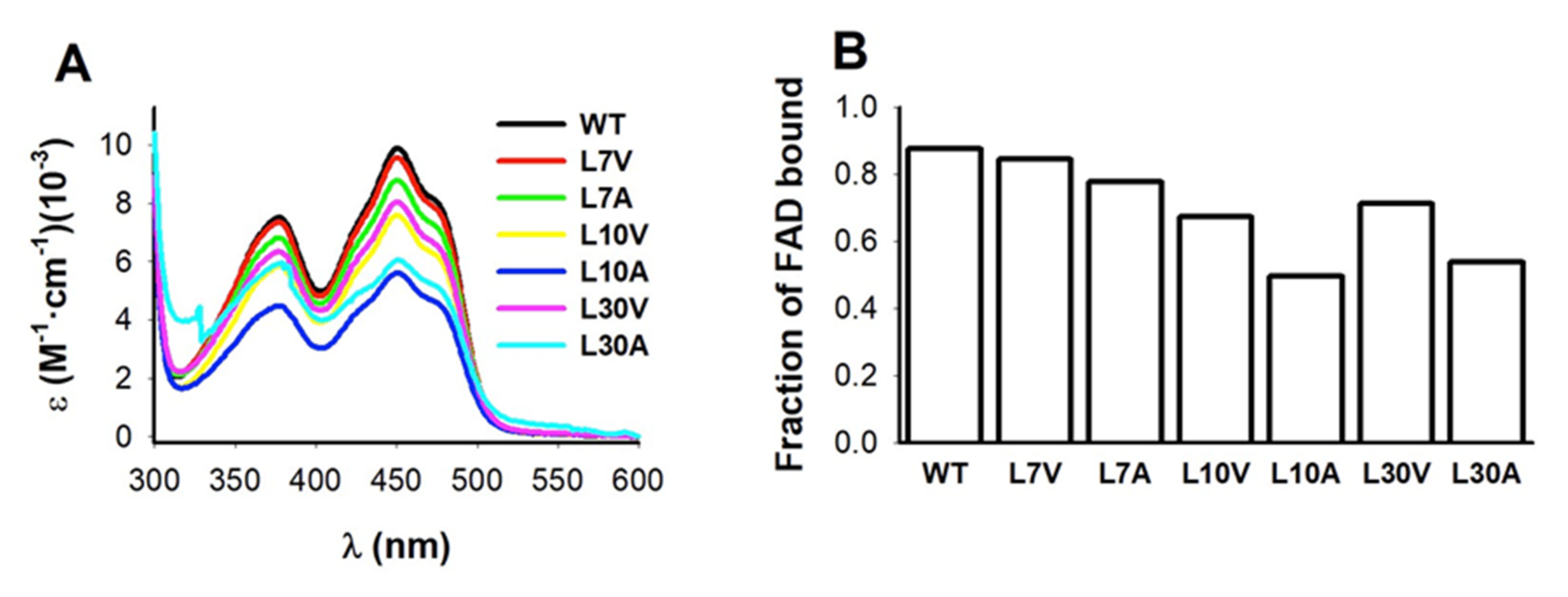
2.4. FAD Content
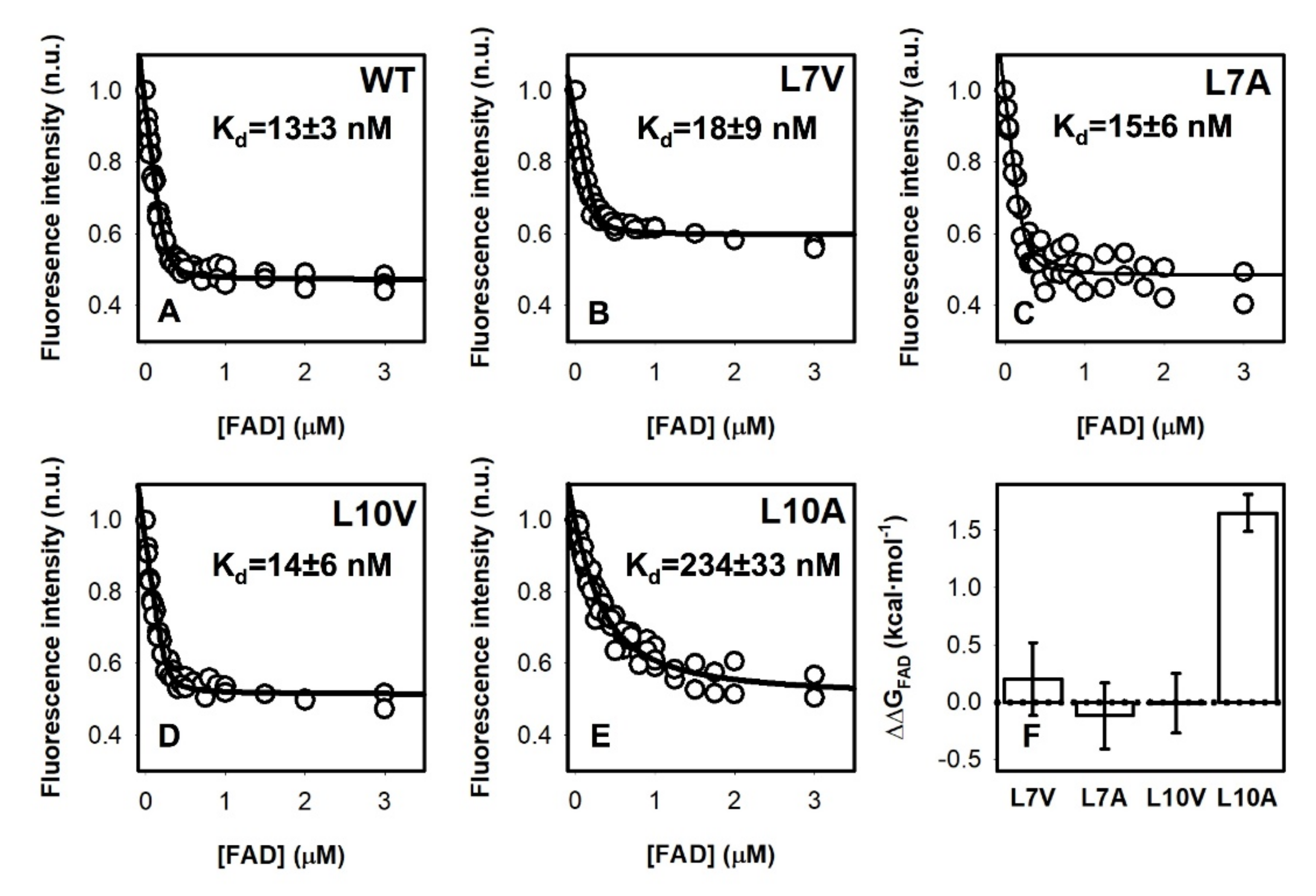
2.5. FAD Binding Affinity
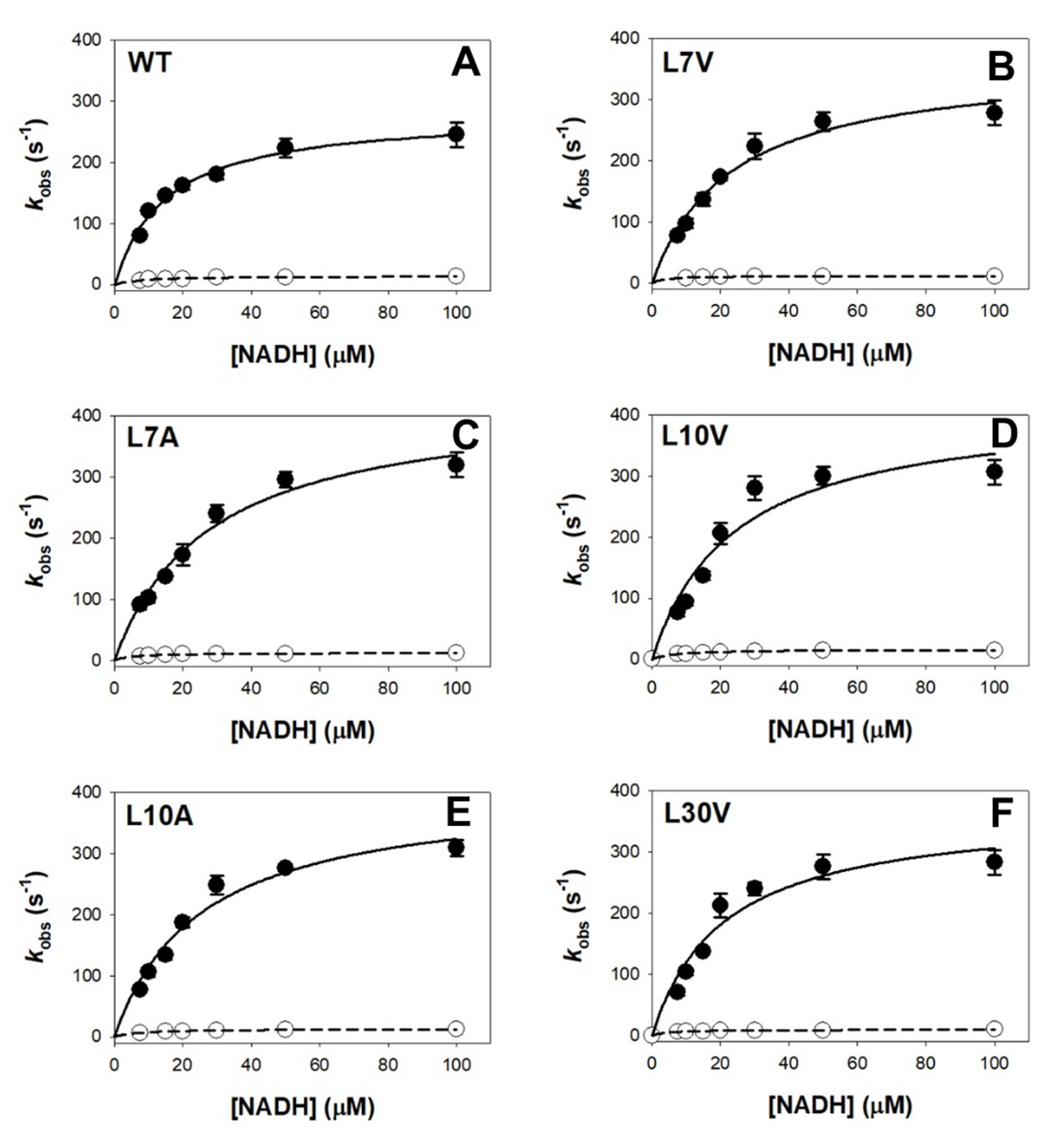
2.6. Enzyme Kinetics for the Reductive Half Reaction with NADH
2.7. Hydrogen/Deuterium Exchange Mass Spectrometry (HDXMS)
2.8. Statistical Mechanical Model Predictions

3. Results and Discussion
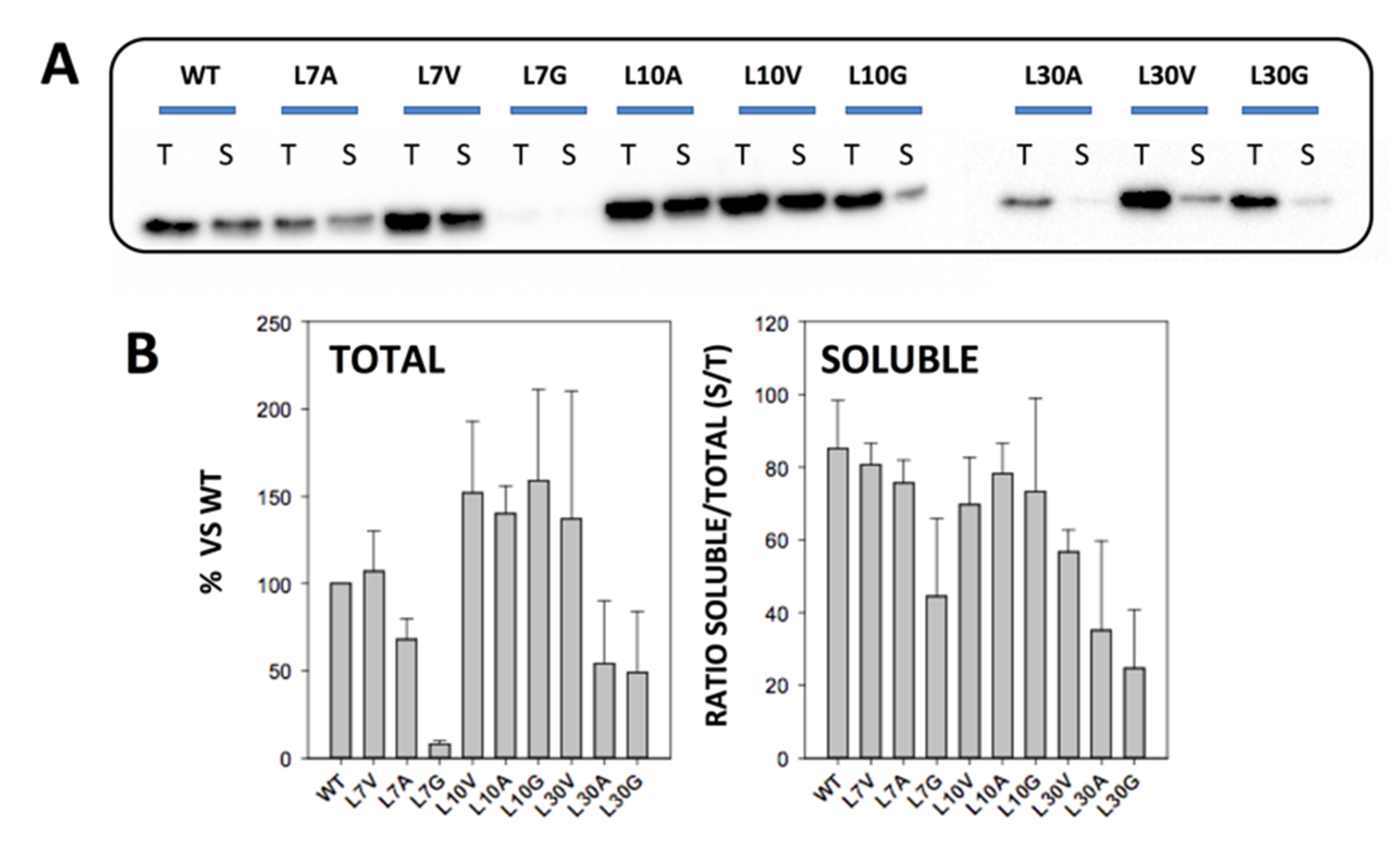
3.1. Expression and Solubility of Cavity-Making Mutants

3.2. Thermal Stability of Cavity-Making Mutants
3.3. Local Stability of the NTD Investigated by Partial Proteolysis with Thermolysin
3.4. FAD Binding Affinity
3.5. Effect of Cavity-Making Mutations on Enzyme Kinetics
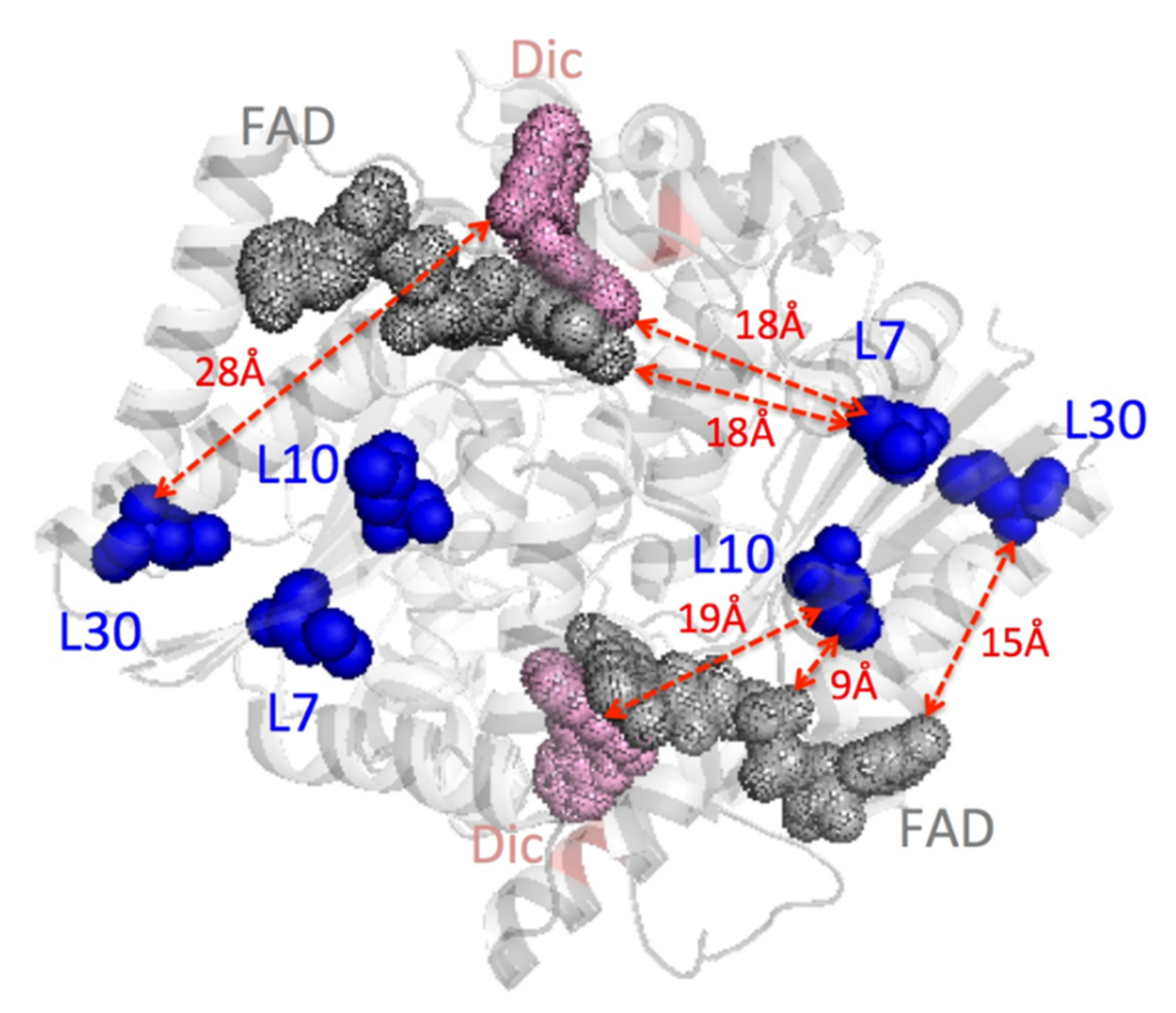
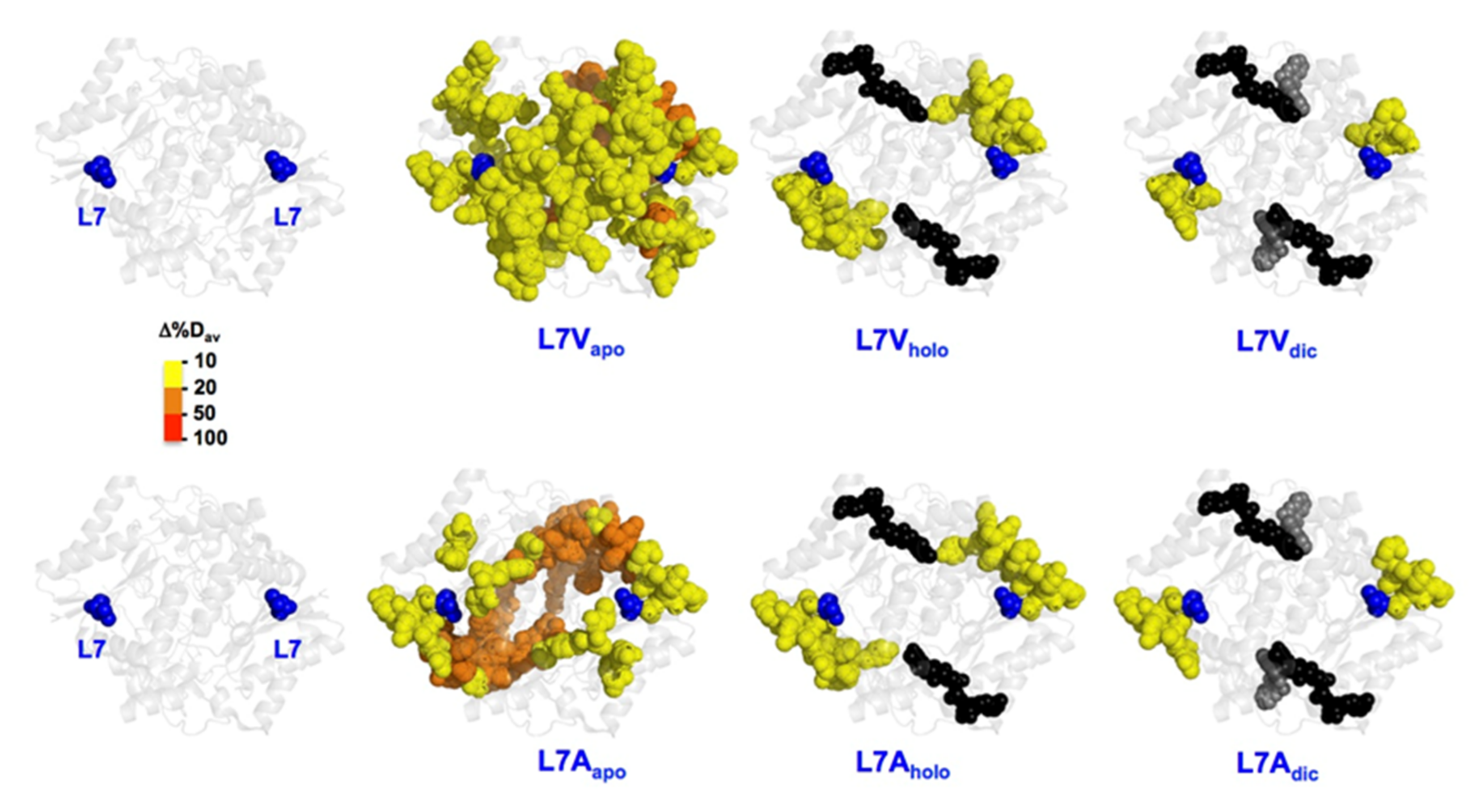
3.6. Effect of Cavity-Making Mutations on the Structural Stability of NQO1
3.6.1. The L7 Cavity-Making Mutants
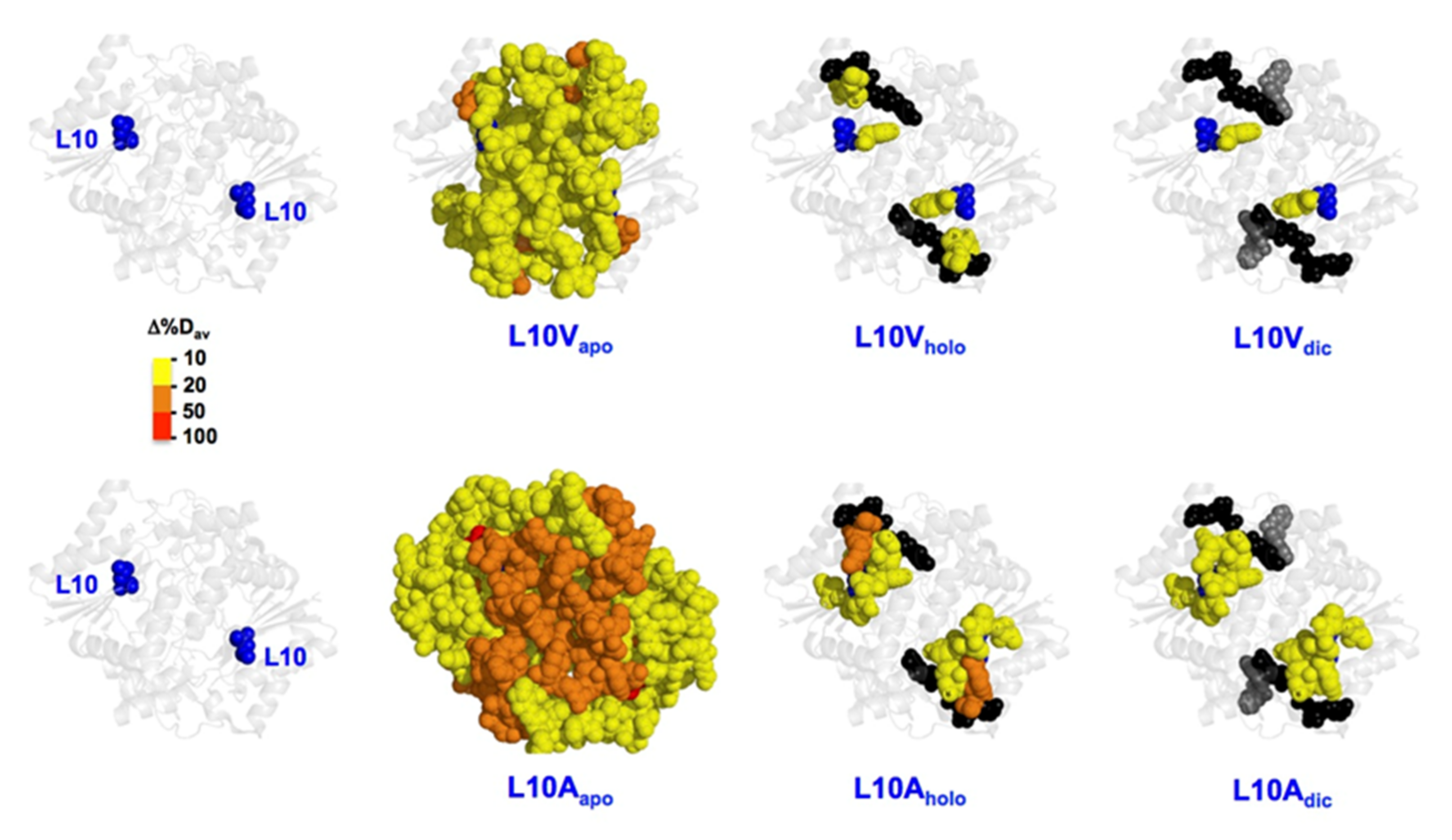
3.6.2. The L10 Cavity-Making Mutants
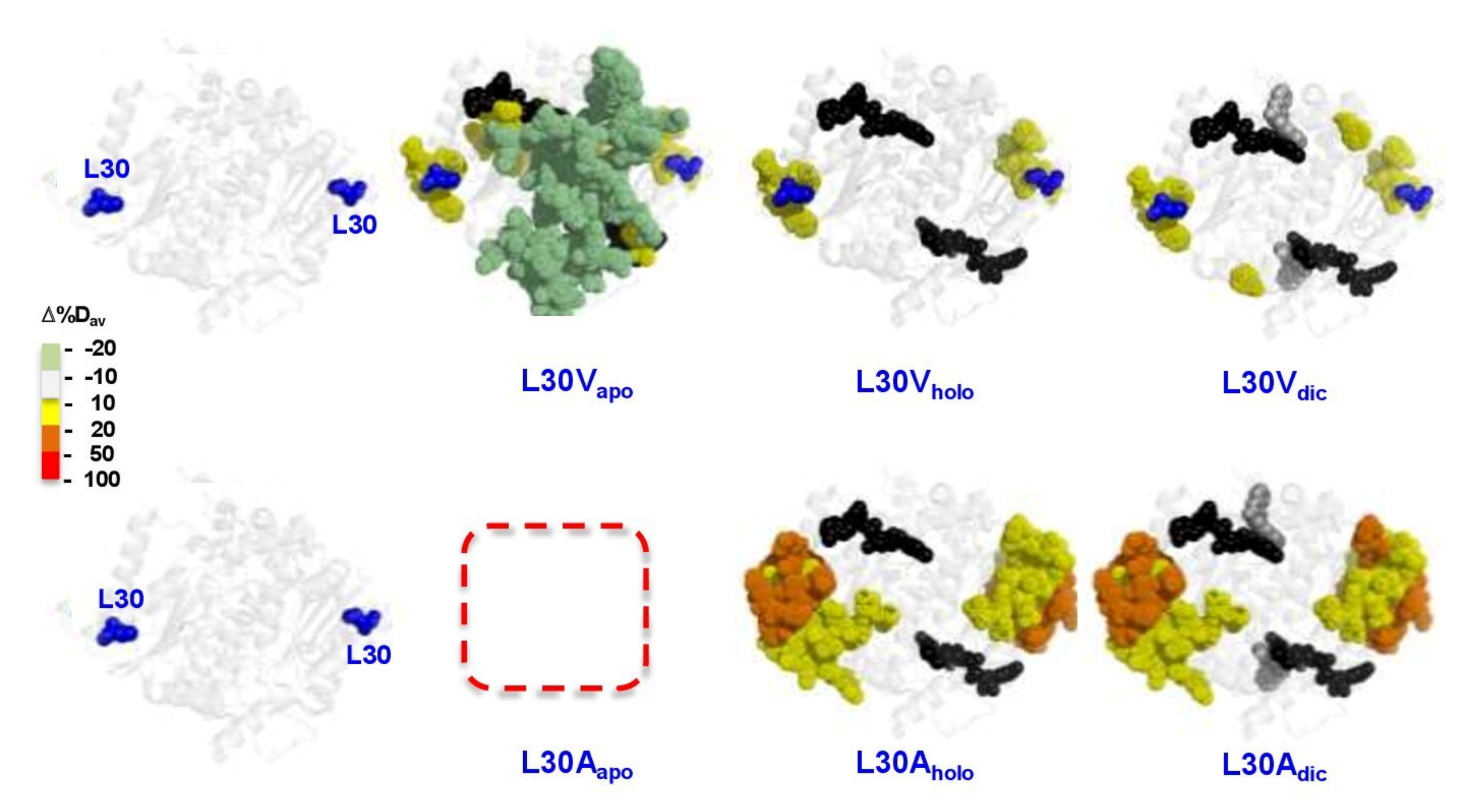
3.6.3. The L30 Cavity-Making Mutants
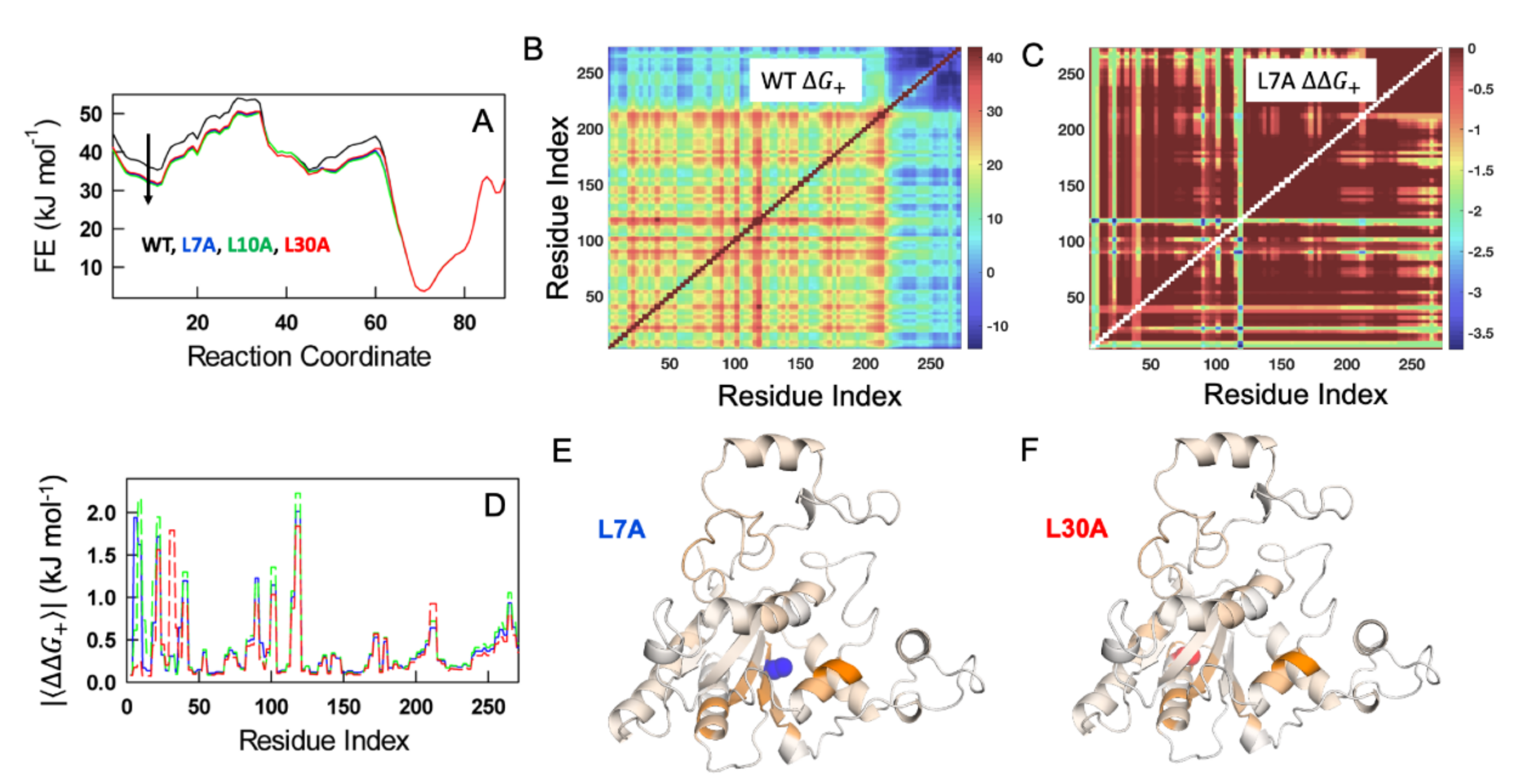
3.7. Statistical Mechanical Calculations on the Effects of NQO1 Cavity-Making Mutants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naganathan, A.N. Modulation of Allosteric Coupling by Mutations: From Protein Dynamics and Packing to Altered Native Ensembles and Function. Curr. Opin. Struct. Biol. 2019, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Garcia, J.L.; Anoz-Carbonell, E.; Vankova, P.; Kannan, A.; Palomino-Morales, R.; Mesa-Torres, N.; Salido, E.; Man, P.; Medina, M.; Naganathan, A.N.; et al. Structural Basis of the Pleiotropic and Specific Phenotypic Consequences of Missense Mutations in the Multifunctional NAD(P)H: Quinone Oxidoreductase 1 and Their Pharmacological Rescue. Redox Biol. 2021, 46, 102112. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.; Suresh, S.; Gopi, S.; Raman, K.; Naganathan, A.N. A General Mechanism for the Propagation of Mutational Effects in Proteins. Biochemistry 2017, 56, 294–305. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Sekhar, A.; Naganathan, A.N. A Universal Pattern in the Percolation and Dissipation of Protein Structural Perturbations. J. Phys. Chem. Lett. 2017, 8, 4779–4784. [Google Scholar] [CrossRef] [PubMed]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A Target for the Treatment of Cancer and Neurological Diseases, and a Model to Understand Loss of Function Disease Mechanisms. Biochim. Biophys. Acta—Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer. Biomolecules 2019, 9, 728. [Google Scholar] [CrossRef] [Green Version]
- Megarity, C.F.; Abdel-Aal Bettley, H.; Caraher, M.C.; Scott, K.A.; Whitehead, R.C.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; et al. Negative Cooperativity in NAD(P)H Quinone Oxidoreductase 1 (NQO1). ChemBioChem 2019, 20, 2841–2849. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H Quinone Oxidoreductase (NQO1): An Enzyme Which Needs Just Enough Mobility, in Just the Right Places. Biosci. Rep. 2019, 39, BSR20180459. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The Diverse Functionality of NQO1 and Its Roles in Redox Control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalayt, P.; Amzel, L.M. The Three-Dimensional Structure of NAD(P)H:Quinone Reductase, a Flavoprotein Involved in Cancer Chemoprotection and Chemotherapy: Mechanism of the Two-Electron. Reduction (x-Ray Diffraction/Flavin). Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [Green Version]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the Native Structure Caused by a Single Amino Acid Exchange in Human NAD(P)H: Quinone Oxidoreductase. FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of Recombinant Human and Mouse NAD(P)H: Quinone Oxidoreductases: Species Comparison and Structural Changes with Substrate Binding and Release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-Site Interdomain Communication May Mediate Different Loss-of-Function Mechanisms in a Cancer-Associated NQO1 Polymorphism. Sci. Rep. 2017, 7, 44532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Encarnación, M.C.; Palomino-Morales, R.J.; Fuchs, J.E.; Esperanza, P.G.; Noel, M.T.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational Dynamics Is Key to Understanding Loss-of-Function of NQO1 Cancer-Associated Polymorphisms and Its Correction by Pharmacological Ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef] [Green Version]
- Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef]
- Xue, M.; Wakamoto, T.; Kejlberg, C.; Yoshimura, Y.; Nielsen, T.A.; Risør, M.W.; Sanggaard, K.W.; Kitahara, R.; Mulder, F.A.A. How Internal Cavities Destabilize a Protein. Proc. Natl. Acad. Sci USA 2019, 116, 21031–21036. [Google Scholar] [CrossRef] [Green Version]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced Vulnerability of Human Proteins towards Disease-Associated Inactivation through Divergent Evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD Binding Overcomes Defects in Activity and Stability Displayed by Cancer-Associated Variants of Human NQO1. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-García, J.L.; Cano-Muñoz, M.; Sánchez-Ramos, I.; Salido, E.; Pey, A.L. Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. J. Pers. Med. 2020, 10, 207. [Google Scholar] [CrossRef]
- Sánchez-Azqueta, A.; Catalano-Dupuy, D.L.; López-Rivero, A.; Tondo, M.L.; Orellano, E.G.; Ceccarelli, E.A.; Medina, M. Dynamics of the Active Site Architecture in Plant-Type Ferredoxin-NADP + Reductases Catalytic Complexes. Biochim. Biophys. Acta—Bioenerg. 2014, 1837, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Trcka, F.; Durech, M.; Man, P.; Hernychova, L.; Muller, P.; Vojtesek, B. The Assembly and Intermolecular Properties of the Hsp70-Tomm34-Hsp90 Molecular Chaperone Complex. J. Biol. Chem. 2014, 289, 9887–9901. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Smith, D.L. Determination of Amide Hydrogen Exchange by Mass Spectrometry: A New Tool for Protein Structure Elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naganathan, A.N.; Dani, R.; Gopi, S.; Aranganathan, A.; Narayan, A. Folding Intermediates, Heterogeneous Native Ensembles and Protein Function. J. Mol. Biol. 2021, 433, 167325. [Google Scholar] [CrossRef]
- Gopi, S.; Aranganathan, A.; Naganathan, A.N. Thermodynamics and Folding Landscapes of Large Proteins from a Statistical Mechanical Model. Curr. Res. Struct. Biol. 2019, 1, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The Crystal Structure of NAD(P)H Quinone Oxidoreductase 1 in Complex with Its Potent Inhibitor Dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Naganathan, A.N.; Kannan, A. A Hierarchy of Coupling Free Energies Underlie the Thermodynamic and Functional Architecture of Protein Structures. Curr. Res. Struct. Biol. 2021, 3, 257–267. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; Schrodinger LLC: New York, NY, USA, 2002. [Google Scholar]
- Pey, A.L. Biophysical and Functional Perturbation Analyses at Cancer-Associated P187 and K240 Sites of the Multifunctional NADP(H): Quinone Oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Klinman, J.P. Update 1 of: Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem. Rev. 2010, 110, PR41–PR67. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | kHTFAST (s−1) | Kd FAST (μM) | kHT/Kd FAST (μM−1·s−1) | kHT SLOW (s−1) | Kd SLOW (μM) | kHT/Kd SLOW (μM−1·s−1) |
|---|---|---|---|---|---|---|
| WT | 281 ± 14 | 15 ± 2 | 19 ± 2 | 14 ± 2 | 8 ± 3 | 1.8 ± 0.6 |
| L7V | 363 ± 26 | 23 ± 4 | 16 ± 2 | 18 ± 1 | 6 ± 1 | 3.0 ± 0.5 |
| L7A | 430 ± 31 | 28 ± 5 | 15 ± 2 | 13 ± 1 | 5 ± 1 | 2.5 ± 0.4 |
| L10V | 417 ± 59 | 24 ± 8 | 17 ± 2 | 16 ± 1 | 7 ± 1 | 2.3 ± 0.8 |
| L10A | 460 ± 30 | 32 ± 5 | 14 ± 2 | 13 ± 1 | 7 ± 1 | 1.8 ± 0.1 |
| L30V | 370 ± 42 | 21 ± 6 | 18 ± 2 | 10 ± 1 | 5 ± 1 | 2.1 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco-Garcia, J.L.; Loginov, D.S.; Anoz-Carbonell, E.; Vankova, P.; Palomino-Morales, R.; Salido, E.; Man, P.; Medina, M.; Naganathan, A.N.; Pey, A.L. Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations. Antioxidants 2022, 11, 1110. https://doi.org/10.3390/antiox11061110
Pacheco-Garcia JL, Loginov DS, Anoz-Carbonell E, Vankova P, Palomino-Morales R, Salido E, Man P, Medina M, Naganathan AN, Pey AL. Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations. Antioxidants. 2022; 11(6):1110. https://doi.org/10.3390/antiox11061110
Chicago/Turabian StylePacheco-Garcia, Juan Luis, Dmitry S. Loginov, Ernesto Anoz-Carbonell, Pavla Vankova, Rogelio Palomino-Morales, Eduardo Salido, Petr Man, Milagros Medina, Athi N. Naganathan, and Angel L. Pey. 2022. "Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations" Antioxidants 11, no. 6: 1110. https://doi.org/10.3390/antiox11061110
APA StylePacheco-Garcia, J. L., Loginov, D. S., Anoz-Carbonell, E., Vankova, P., Palomino-Morales, R., Salido, E., Man, P., Medina, M., Naganathan, A. N., & Pey, A. L. (2022). Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations. Antioxidants, 11(6), 1110. https://doi.org/10.3390/antiox11061110