
Antioxidant Therapy in Cancer: Rationale and Progress

 , ,
, , 
Abstract
:1. Introduction
2. Redox Homeostasis: The Biological Basis for Antioxidant Therapy
2.1. Mechanisms in ROS Generation
2.2. ROS Elimination with Enzymatic or Nonenzymatic Antioxidant System
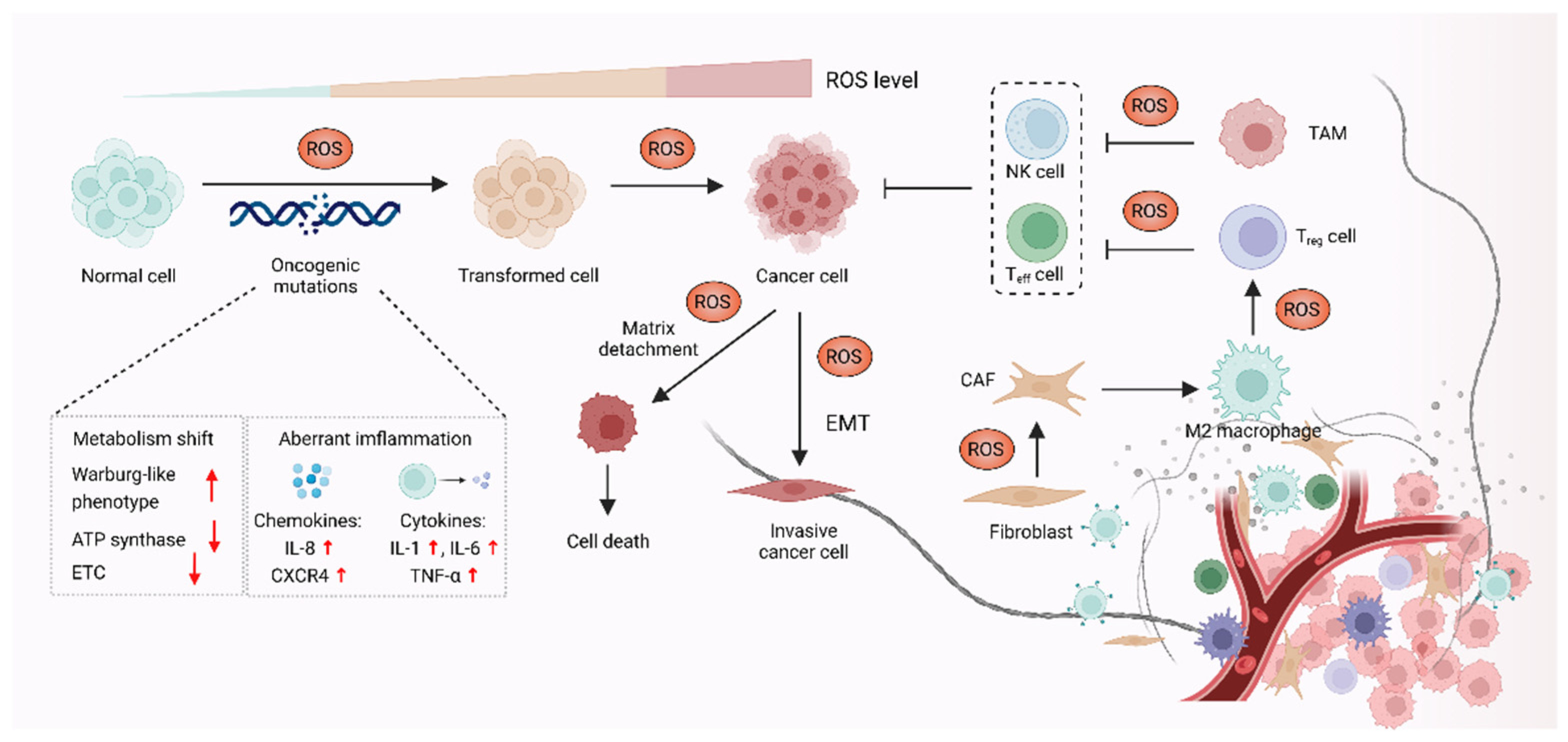
3. ROS Promote Carcinogenesis and Cancer Progression
3.1. ROS-Mediated Oncogenic Mutations Promote Carcinogenesis
3.2. ROS Function as Signaling Molecules to Drive Cancer Progression
4. Antioxidant Therapeutic Strategies in Cancer
4.1. Targeting ROS with Nonenzymatic Antioxidants
4.2. Targeting ROS with Enzymatic Antioxidants
5. Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jiang, J.; Chen, H.N.; Zhou, L.; Huang, Z.; Qin, S.; Jin, P.; Luo, M.; Li, B.; Shi, J.; et al. Redox-sensitive cyclophilin A elicits chemoresistance through realigning cellular oxidative status in colorectal cancer. Cell Rep. 2021, 37, 110069. [Google Scholar] [CrossRef]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic-Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019, 44, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Jaen, R.I.; Regdon, Z.; Bosca, L.; Prieto, P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. 2019, 26, 101261. [Google Scholar] [CrossRef] [PubMed]
- Caserta, S.; Ghezzi, P. Release of redox enzymes and micro-RNAs in extracellular vesicles, during infection and inflammation. Free Radic. Biol. Med. 2021, 169, 248–257. [Google Scholar] [CrossRef]
- Gonzalez-Chavez, Z.; Vazquez, C.; Mejia-Tlachi, M.; Marquez-Duenas, C.; Manning-Cela, R.; Encalada, R.; Rodriguez-Enriquez, S.; Michels, P.A.M.; Moreno-Sanchez, R.; Saavedra, E. Gamma-glutamylcysteine synthetase and tryparedoxin 1 exert high control on the antioxidant system in Trypanosoma cruzi contributing to drug resistance and infectivity. Redox Biol. 2019, 26, 101231. [Google Scholar] [CrossRef]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef]
- Urso, L.; Cavallari, I.; Sharova, E.; Ciccarese, F.; Pasello, G.; Ciminale, V. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. Br. J. Cancer 2020, 122, 52–61. [Google Scholar] [CrossRef]
- Canli, O.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Lower, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Huang, Z.; Yang, X.; Chen, Y.; Jiang, J.; Zhang, L.; Zhou, L.; Qin, S.; Jin, P.; Fu, S.; et al. PHLDB2 Mediates Cetuximab Resistance via Interacting With EGFR in Latent Metastasis of Colorectal Cancer. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1223–1242. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef]
- Li, P.; Wu, M.; Wang, J.; Sui, Y.; Liu, S.; Shi, D. NAC selectively inhibit cancer telomerase activity: A higher redox homeostasis threshold exists in cancer cells. Redox Biol. 2016, 8, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox Biol. 2019, 25, 101139. [Google Scholar] [CrossRef]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Q.; Cai, J.; Wang, Y.; Lai, X.; Qiu, Y.; Huang, Y.; Ke, Q.; Zhang, Y.; Guan, Y.; et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nat. Commun. 2019, 10, 5043. [Google Scholar] [CrossRef] [Green Version]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Cantoni, O.; Zito, E.; Fiorani, M.; Guidarelli, A. Arsenite impinges on endoplasmic reticulum-mitochondria crosstalk to elicit mitochondrial ROS formation and downstream toxicity. Semin. Cancer Biol. 2021, 76, 132–138. [Google Scholar] [CrossRef]
- He, A.; Dean, J.M.; Lodhi, I.J. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol. 2021, 31, 656–670. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Flockhart, M.; Nilsson, L.C.; Tais, S.; Ekblom, B.; Apro, W.; Larsen, F.J. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. 2021, 33, 957–970.e956. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, R.; Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Sobrevia, L. Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165961. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.; Rolas, L.; El-Benna, J. The Dual Role of Reactive Oxygen Species-Generating Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Gastrointestinal Inflammation and Therapeutic Perspectives. Antioxid. Redox Signal. 2020, 33, 354–373. [Google Scholar] [CrossRef]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef]
- Wadgaonkar, P.; Chen, F. Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. 2021, 76, 258–266. [Google Scholar] [CrossRef]
- Bibli, S.I.; Fleming, I. Oxidative Post-Translational Modifications: A Focus on Cysteine S-Sulfhydration and the Regulation of Endothelial Fitness. Antioxid. Redox Signal. 2021, 35, 1494–1514. [Google Scholar] [CrossRef]
- Samanta, S.; Yang, S.; Debnath, B.; Xue, D.; Kuang, Y.; Ramkumar, K.; Lee, A.S.; Ljungman, M.; Neamati, N. The Hydroxyquinoline Analogue YUM70 Inhibits GRP78 to Induce ER Stress-Mediated Apoptosis in Pancreatic Cancer. Cancer Res. 2021, 81, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020, 30, 672–675. [Google Scholar] [CrossRef]
- Wong, C.O.; Karagas, N.E.; Jung, J.; Wang, Q.; Rousseau, M.A.; Chao, Y.; Insolera, R.; Soppina, P.; Collins, C.A.; Zhou, Y.; et al. Regulation of longevity by depolarization-induced activation of PLC-beta-IP3R signaling in neurons. Proc. Natl. Acad. Sci. USA 2021, 118, e2004253118. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 2021, 81, 3866–3876.e3862. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C. Redox Signaling from and to Peroxisomes: Progress, Challenges, and Prospects. Antioxid. Redox Signal. 2019, 30, 95–112. [Google Scholar] [CrossRef]
- Sargsyan, Y.; Thoms, S. Staying in Healthy Contact: How Peroxisomes Interact with Other Cell Organelles. Trends Mol. Med. 2020, 26, 201–214. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.L.; Wangler, M.F.; Marcogliese, P.C.; Jo, J.; Ravenscroft, T.A.; Zuo, Z.; Duraine, L.; Sadeghzadeh, S.; Li-Kroeger, D.; Schmidt, R.E.; et al. Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms. Neuron 2020, 106, 589–606.e586. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Nishigori, C.; Hattori, Y.; Toyokuni, S. Role of reactive oxygen species in skin carcinogenesis. Antioxid. Redox Signal. 2004, 6, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Allouche, J.; Rachmin, I.; Adhikari, K.; Pardo, L.M.; Lee, J.H.; McConnell, A.M.; Kato, S.; Fan, S.; Kawakami, A.; Suita, Y.; et al. NNT mediates redox-dependent pigmentation via a UVB- and MITF-independent mechanism. Cell 2021, 184, 4268–4283.e4220. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Maddirela, D.; Banasavadi-Siddegowda, Y.K.; Lai, T.H.; Qamri, Z.; Jacob, N.K.; Sampath, D.; Mohanam, S.; Kaur, B.; Puduvalli, V.K. A novel interaction of PAK4 with PPARgamma to regulate Nox1 and radiation-induced epithelial-to-mesenchymal transition in glioma. Oncogene 2017, 36, 5309–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Shui, S.; Zhao, Z.; Wang, H.; Conrad, M.; Liu, G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021, 45, 102056. [Google Scholar] [CrossRef]
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Vo, T.N.; Ezerina, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959. [Google Scholar] [CrossRef]
- Patgiri, A.; Skinner, O.S.; Miyazaki, Y.; Schleifer, G.; Marutani, E.; Shah, H.; Sharma, R.; Goodman, R.P.; To, T.L.; Robert Bao, X.; et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat. Biotechnol. 2020, 38, 309–313. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jimenez-Altayo, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2021, 162, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montllor-Albalate, C.; Colin, A.E.; Chandrasekharan, B.; Bolaji, N.; Andersen, J.L.; Wayne Outten, F.; Reddi, A.R. Extra-mitochondrial Cu/Zn superoxide dismutase (Sod1) is dispensable for protection against oxidative stress but mediates peroxide signaling in Saccharomyces cerevisiae. Redox Biol. 2019, 21, 101064. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Cobine, P.A.; Molik, S.; Naranuntarat, A.; Lill, R.; Winge, D.R.; Culotta, V.C. The effects of mitochondrial iron homeostasis on cofactor specificity of superoxide dismutase 2. EMBO J. 2006, 25, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Mohammedi, K.; Bellili-Munoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Dong, Y.; Liu, B.; Liu, J.; Liu, S.; Zhao, Z.; Li, W.; Tian, B.; Zhao, R.; He, F.; et al. Guiding Transition Metal-Doped Hollow Cerium Tandem Nanozymes with Elaborately Regulated Multi-Enzymatic Activities for Intensive Chemodynamic Therapy. Adv. Mater. 2022, 34, e2107054. [Google Scholar] [CrossRef]
- Robbins, M.E.; Cho, H.Y.; Hansen, J.M.; Luchsinger, J.R.; Locy, M.L.; Velten, M.; Kleeberger, S.R.; Rogers, L.K.; Tipple, T.E. Glutathione reductase deficiency alters lung development and hyperoxic responses in neonatal mice. Redox Biol. 2021, 38, 101797. [Google Scholar] [CrossRef]
- Luzzatto, L.; Arese, P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N. Engl. J. Med. 2018, 378, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef]
- Fumarola, S.; Cecati, M.; Sartini, D.; Ferretti, G.; Milanese, G.; Galosi, A.B.; Pozzi, V.; Campagna, R.; Morresi, C.; Emanuelli, M.; et al. Bladder Cancer Chemosensitivity is Affected by Paraoxonase-2 Expression. Antioxidants 2020, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Campagna, R.; Bacchetti, T.; Salvolini, E.; Pozzi, V.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Ferretti, G.; Offidani, A.; et al. Paraoxonase-2 Silencing Enhances Sensitivity of A375 Melanoma Cells to Treatment with Cisplatin. Antioxidants 2020, 9, 1238. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, T.; Song, J.; Wang, Y.; Guo, L.; Yuan, L.; Zhao, Y.; Gao, Y.; Lin, L.; Wang, Y.; Wei, J. Expression and characterization of recombinant bifunctional enzymes with glutathione peroxidase and superoxide dismutase activities. Free Radic. Biol. Med. 2017, 110, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wu, G.; Zhu, J.; Tan, Z.; Shi, D.; Wu, X.; Tang, M.; Li, Z.; Hu, Y.; Zhang, S.; et al. Genotoxic stress-triggered beta-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis. Nat. Commun. 2019, 10, 3761. [Google Scholar] [CrossRef] [PubMed]
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, C.M.; Botta, D.; White, C.C.; Scoville, D.K.; Srinouanprachanh, S.; Bammler, T.K.; MacDonald, J.; Kavanagh, T.J. Persistence of improved glucose homeostasis in Gclm null mice with age and cadmium treatment. Redox Biol. 2022, 49, 102213. [Google Scholar] [CrossRef]
- Kelly-Aubert, M.; Trudel, S.; Fritsch, J.; Nguyen-Khoa, T.; Baudouin-Legros, M.; Moriceau, S.; Jeanson, L.; Djouadi, F.; Matar, C.; Conti, M.; et al. GSH monoethyl ester rescues mitochondrial defects in cystic fibrosis models. Hum. Mol. Genet. 2011, 20, 2745–2759. [Google Scholar] [CrossRef] [Green Version]
- Navarro, J.; Obrador, E.; Carretero, J.; Petschen, I.; Avino, J.; Perez, P.; Estrela, J.M. Changes in glutathione status and the antioxidant system in blood and in cancer cells associate with tumour growth in vivo. Free Radic. Biol. Med. 1999, 26, 410–418. [Google Scholar] [CrossRef]
- McCarver, A.C.; Lessner, D.J. Molecular characterization of the thioredoxin system from Methanosarcina acetivorans. FEBS J. 2014, 281, 4598–4611. [Google Scholar] [CrossRef] [Green Version]
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of beta-cell function by thioredoxin. Redox Biol. 2020, 34, 101570. [Google Scholar] [CrossRef]
- Peskin, A.V.; Meotti, F.C.; Kean, K.M.; Gobl, C.; Peixoto, A.S.; Pace, P.E.; Horne, C.R.; Heath, S.G.; Crowther, J.M.; Dobson, R.C.J.; et al. Modifying the resolving cysteine affects the structure and hydrogen peroxide reactivity of peroxiredoxin 2. J. Biol. Chem. 2021, 296, 100494. [Google Scholar] [CrossRef] [PubMed]
- Perez-Jimenez, R.; Li, J.; Kosuri, P.; Sanchez-Romero, I.; Wiita, A.P.; Rodriguez-Larrea, D.; Chueca, A.; Holmgren, A.; Miranda-Vizuete, A.; Becker, K.; et al. Diversity of chemical mechanisms in thioredoxin catalysis revealed by single-molecule force spectroscopy. Nat. Struct. Mol. Biol. 2009, 16, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Biophys. Acta 2015, 1850, 1607–1621. [Google Scholar] [CrossRef] [Green Version]
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1–3 Signal Transduction Pathways. Antioxid. Redox Signal. 2019, 30, 443–486. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Koelwyn, G.J.; Quail, D.F.; Zhang, X.; White, R.M.; Jones, L.W. Exercise-dependent regulation of the tumour microenvironment. Nat. Rev. Cancer 2017, 17, 620–632. [Google Scholar] [CrossRef]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Gobl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 2015, 35, S199–S223. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting MARCO and IL37R on Immunosuppressive Macrophages in Lung Cancer Blocks Regulatory T Cells and Supports Cytotoxic Lymphocyte Function. Cancer Res. 2021, 81, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.N.; Shirmanova, M.V.; Lukina, M.M.; Dudenkova, V.V.; Mishina, N.M.; Zagaynova, E.V. The metabolic interaction of cancer cells and fibroblasts—coupling between NAD(P)H and FAD, intracellular pH and hydrogen peroxide. Cell Cycle 2016, 15, 1257–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Kastl, L.; Sauer, S.; Beissbarth, T.; Becker, M.; Krammer, P.; Gulow, K. TNF-a stimulation enhances ROS-dependent cell migration via NF-?B activation in liver cells. Free Radic. Biol. Med. 2014, 75 (Suppl. S1), S32. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036.e1029. [Google Scholar] [CrossRef] [Green Version]
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Schmidlin, C.J.; Shakya, A.; Dodson, M.; Chapman, E.; Zhang, D.D. The intricacies of NRF2 regulation in cancer. Semin. Cancer Biol. 2021, 76, 110–119. [Google Scholar] [CrossRef]
- Young, M.R.I.; Xiong, Y. Influence of vitamin D on cancer risk and treatment: Why the variability? Trends Cancer Res. 2018, 13, 43–53. [Google Scholar]
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New potential biomarker for stratification of patients for pharmacological vitamin C in adjuvant settings of cancer therapy. Redox Biol. 2020, 28, 101357. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury. Antioxid. Redox Signal. 2018, 29, 1691–1724. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Y.; Kim, H.J.; Lee, J.W.; Chung, I.Y.; Kim, J.S.; Lee, S.B.; Son, B.H.; Eom, J.S.; Kim, S.B.; Gong, G.Y.; et al. Breast Cancer Recurrence in the Nipple-Areola Complex After Nipple-Sparing Mastectomy With Immediate Breast Reconstruction for Invasive Breast Cancer. JAMA Surg. 2019, 154, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Batist, G. Glutathione and glutathione analogues; therapeutic potentials. Biochim. Biophys. Acta 2013, 1830, 3350–3353. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Chun, K.S.; Kim, D.H.; Kim, S.J.; Kim, S.H.; Cho, N.C.; Na, H.K.; Surh, Y.J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cocheme, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Covas, G.; Marinho, H.S.; Cyrne, L.; Antunes, F. Activation of Nrf2 by H2O2: De novo synthesis versus nuclear translocation. Methods Enzymol. 2013, 528, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Casares, L.; Unciti-Broceta, J.D.; Prados, M.E.; Caprioglio, D.; Mattoteia, D.; Higgins, M.; Apendino, G.; Dinkova-Kostova, A.T.; Munoz, E.; de la Vega, L. Isomeric O-methyl cannabidiolquinones with dual BACH1/NRF2 activity. Redox Biol. 2020, 37, 101689. [Google Scholar] [CrossRef]
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The Pharmacokinetics of Fumaric Acid Esters Reveal Their In Vivo Effects. Trends Pharmacol. Sci. 2018, 39, 1–12. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [Green Version]
- Galloway, D.A.; Williams, J.B.; Moore, C.S. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann. Clin. Transl. Neurol. 2017, 4, 381–391. [Google Scholar] [CrossRef]
- Higashi, C.; Kawaji, A.; Tsuda, N.; Hayashi, M.; Saito, R.; Yagishita, Y.; Suzuki, T.; Uruno, A.; Nakamura, M.; Nakao, K.; et al. The novel Nrf2 inducer TFM-735 ameliorates experimental autoimmune encephalomyelitis in mice. Eur. J. Pharmacol. 2017, 802, 76–84. [Google Scholar] [CrossRef]
- Delmastro-Greenwood, M.; Hughan, K.S.; Vitturi, D.A.; Salvatore, S.R.; Grimes, G.; Potti, G.; Shiva, S.; Schopfer, F.J.; Gladwin, M.T.; Freeman, B.A.; et al. Nitrite and nitrate-dependent generation of anti-inflammatory fatty acid nitroalkenes. Free Radic. Biol. Med. 2015, 89, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.C.; Zhao, J.; Liu, Y.T.; Liu, T.; Tao, M.M.; You, Q.D.; Jiang, Z.Y. CPUY192018, a potent inhibitor of the Keap1-Nrf2 protein-protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF-kappaB activation. Redox Biol. 2019, 26, 101266. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.C.; Ji, J.A.; Jiang, Y.L.; Chen, Z.Y.; Yuan, Z.W.; You, Q.D.; Jiang, Z.Y. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 2016, 6, 26585. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329e.318. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663. [Google Scholar] [CrossRef]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Pilewskie, M.; Morrow, M. Axillary Nodal Management Following Neoadjuvant Chemotherapy: A Review. JAMA Oncol. 2017, 3, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacciatore, I.; Cornacchia, C.; Pinnen, F.; Mollica, A.; Di Stefano, A. Prodrug approach for increasing cellular glutathione levels. Molecules 2010, 15, 1242–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, R.N.; Meister, A. Transport of glutathione, as gamma-glutamylcysteinylglycyl ester, into liver and kidney. Proc. Natl. Acad. Sci. USA 1983, 80, 5258–5260. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Neto, J.C.; Round, J.L. Gut microbiota: A new way to take your vitamins. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 521–522. [Google Scholar] [CrossRef]
- Said, H.M.; Nexo, E. Gastrointestinal Handling of Water-Soluble Vitamins. Compr. Physiol. 2018, 8, 1291–1311. [Google Scholar] [CrossRef]
- Cossey, L.N.; Rahim, F.; Larsen, C.P. Oxalate nephropathy and intravenous vitamin C. Am. J. Kidney Dis. 2013, 61, 1032–1035. [Google Scholar] [CrossRef]
- Mikirova, N.; Casciari, J.; Rogers, A.; Taylor, P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J. Transl. Med. 2012, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer: A meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231. [Google Scholar] [CrossRef]
- Carr, A.C.; Cook, J. Intravenous Vitamin C for Cancer Therapy—Identifying the Current Gaps in Our Knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [Google Scholar] [CrossRef]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sauberlich, H.E. Pharmacology of vitamin C. Annu. Rev. Nutr. 1994, 14, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Suh, N.; Kong, A.N. Does vitamin E prevent or promote cancer? Cancer Prev. Res. 2012, 5, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal. 2020, 33, 435–454. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox Inhibitors & Therapies: Rational Design of Peptidic and Small Molecule Inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Pagano, E.; Csanyi, G.; Pagano, P.J. NADPH oxidase inhibitors: A decade of discovery from Nox2ds to HTS. Cell Mol. Life Sci. 2012, 69, 2315–2325. [Google Scholar] [CrossRef] [Green Version]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- DiStasi, M.R.; Mund, J.A.; Bohlen, H.G.; Miller, S.J.; Ingram, D.A.; Dalsing, M.C.; Unthank, J.L. Impaired compensation to femoral artery ligation in diet-induced obese mice is primarily mediated via suppression of collateral growth by Nox2 and p47phox. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1207–H1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krotz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.; Li, M.W.; Lemarie, C.A.; Simeone, S.M.; Pagano, P.J.; Gaestel, M.; Paradis, P.; Wassmann, S.; Schiffrin, E.L. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: Regulation of oxidative stress. Hypertension 2011, 57, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Zhang, L.; Hong, J.S.; Zhang, D.; Zhao, J.; Wang, Q. Nicotinamide Adenine Dinucleotide Phosphate Oxidase and Neurodegenerative Diseases: Mechanisms and Therapy. Antioxid. Redox Signal. 2020, 33, 374–393. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H.; Gaur, S.; Markel, S.; Lu, J.; van Balgooy, J.; Synold, T.W.; Xi, B.; Wu, X.; Juhasz, A. Effects of iodonium-class flavin dehydrogenase inhibitors on growth, reactive oxygen production, cell cycle progression, NADPH oxidase 1 levels, and gene expression in human colon cancer cells and xenografts. Free Radic. Biol. Med. 2013, 57, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K. NADPH oxidases: Current aspects and tools. Redox Biol. 2020, 34, 101512. [Google Scholar] [CrossRef]
- Mora-Pale, M.; Kwon, S.J.; Linhardt, R.J.; Dordick, J.S. Trimer hydroxylated quinone derived from apocynin targets cysteine residues of p47phox preventing the activation of human vascular NADPH oxidase. Free Radic. Biol. Med. 2012, 52, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Murley, J.S.; Arbiser, J.L.; Weichselbaum, R.R.; Grdina, D.J. ROS modifiers and NOX4 affect the expression of the survivin-associated radio-adaptive response. Free Radic. Biol. Med. 2018, 123, 39–52. [Google Scholar] [CrossRef]
- Bhandarkar, S.S.; Jaconi, M.; Fried, L.E.; Bonner, M.Y.; Lefkove, B.; Govindarajan, B.; Perry, B.N.; Parhar, R.; Mackelfresh, J.; Sohn, A.; et al. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Investig. 2009, 119, 2359–2365. [Google Scholar] [CrossRef]
- Cao, F.; Zhang, L.; You, Y.; Zheng, L.; Ren, J.; Qu, X. An Enzyme-Mimicking Single-Atom Catalyst as an Efficient Multiple Reactive Oxygen and Nitrogen Species Scavenger for Sepsis Management. Angew. Chem. Int. Ed. Engl. 2020, 59, 5108–5115. [Google Scholar] [CrossRef]
- Carillon, J.; Rouanet, J.M.; Cristol, J.P.; Brion, R. Superoxide dismutase administration, a potential therapy against oxidative stress related diseases: Several routes of supplementation and proposal of an original mechanism of action. Pharm. Res. 2013, 30, 2718–2728. [Google Scholar] [CrossRef] [PubMed]
- Johnke, R.M.; Sattler, J.A.; Allison, R.R. Radioprotective agents for radiation therapy: Future trends. Future Oncol. 2014, 10, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Weitner, T.; Kos, I.; Sheng, H.; Tovmasyan, A.; Reboucas, J.S.; Fan, P.; Warner, D.S.; Vujaskovic, Z.; Batinic-Haberle, I.; Spasojevic, I. Comprehensive pharmacokinetic studies and oral bioavailability of two Mn porphyrin-based SOD mimics, MnTE-2-PyP5+ and MnTnHex-2-PyP5+. Free Radic. Biol. Med. 2013, 58, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovmasyan, A.; Reboucas, J.S.; Benov, L. Simple biological systems for assessing the activity of superoxide dismutase mimics. Antioxid. Redox Signal. 2014, 20, 2416–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chemistry 2018, 24, 5032–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherlock, L.G.; Trumpie, A.; Hernandez-Lagunas, L.; McKenna, S.; Fisher, S.; Bowler, R.; Wright, C.J.; Delaney, C.; Nozik-Grayck, E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants 2018, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Zhu, Y.; Tong, Q.; Kosmacek, E.A.; Lichter, E.Z.; Oberley-Deegan, R.E. The Addition of Manganese Porphyrins during Radiation Inhibits Prostate Cancer Growth and Simultaneously Protects Normal Prostate Tissue from Radiation Damage. Antioxidants 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I. SOD therapeutics: Latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid. Redox Signal. 2014, 20, 2372–2415. [Google Scholar] [CrossRef]
- Sishc, B.J.; Ding, L.; Nam, T.K.; Heer, C.D.; Rodman, S.N.; Schoenfeld, J.D.; Fath, M.A.; Saha, D.; Pulliam, C.F.; Langen, B.; et al. Avasopasem manganese synergizes with hypofractionated radiation to ablate tumors through the generation of hydrogen peroxide. Sci. Transl. Med. 2021, 13, eabb3768. [Google Scholar] [CrossRef]
- Heer, C.D.; Davis, A.B.; Riffe, D.B.; Wagner, B.A.; Falls, K.C.; Allen, B.G.; Buettner, G.R.; Beardsley, R.A.; Riley, D.P.; Spitz, D.R. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H(2)O(2)-Dependent Manner. Antioxidants 2018, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- El-Mahdy, M.A.; Alzarie, Y.A.; Hemann, C.; Badary, O.A.; Nofal, S.; Zweier, J.L. The novel SOD mimetic GC4419 increases cancer cell killing with sensitization to ionizing radiation while protecting normal cells. Free Radic. Biol. Med. 2020, 160, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Asadi, Z.; Mandegani, Z.; Asadi, M.; Pakiari, A.H.; Salarhaji, M.; Manassir, M.; Karbalaei-Heidari, H.R.; Rastegari, B.; Sedaghat, M. Substituted effect on some water-soluble Mn(II) salen complexes: DNA binding, cytotoxicity, molecular docking, DFT studies and theoretical IR & UV studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 206, 278–294. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidants | Cancer Types | Trial Status | Trial ID |
|---|---|---|---|
| NRF2 activators | |||
| Sulforaphane | Lung cancer | Phase 2 | NCT03232138 |
| Breast cancer | Phase 2 | NCT00982319 | |
| Prostate cancer | Phase 2 | NCT01228084 | |
| Colon cancer | NA | NCT01344330 | |
| HNSCC | Early Phase 1 | NCT03182959 | |
| Resveratrol | Colon cancer | Phase 1 | NCT00256334 |
| Colorectal cancer | Phase 1 | NCT00920803 | |
| Neuroendocrine tumor | NA | NCT01476592 | |
| Breast cancer | NA | NCT03482401 | |
| Multiple myeloma | Phase 2 | NCT00920556 | |
| Quercetin | Prostate cancer | Phase 1 | NCT01912820 |
| Colorectal cancer | NA | NCT00003365 | |
| Pancreatic cancer/NSCLC | Phase 2/3 | NCT02195232 | |
| Curcumin | Breast cancer | Phase 2 | NCT01042938 |
| Colorectal cancer | Phase 2 | NCT02439385 | |
| Prostate cancer | NA | NCT03211104 | |
| Head and neck cancer | Early Phase 1 | NCT01160302 | |
| Pancreatic cancer | Phase 2 | NCT00192842 | |
| Bardoxolone-methyl (CDDO-Me, RTA402) | Solid tumors/Lymphoid malignancies | Phase 1 | NCT00529438 |
| Pancreatic cancer | Phase 1 | NCT00529113 | |
| Solid tumors/ Lymphoid malignancies | Phase 1 | NCT00508807 | |
| RTA-408 (omaveloxolone) | NSCLC | Phase 1 | NCT02029729 |
| Breast cancer | Phase 2 | NCT02142959 | |
| Melanoma | Phase 1/2 | NCT02259231 | |
| Dimethyl fumarate | Multiple sclerosis | Phase 3 | NCT02430532 |
| Lymphocytic leukemia | Phase 1 | NCT02784834 | |
| Glioblastoma | Phase 1 | NCT02337426 | |
| Oltipraz | Lung cancer | Phase 1 | NCT00006457 |
| SOD mimics | |||
| GC4419 | Head and neck cancer | Phase 2 | NCT04529850 |
| Pancreatic cancer | Phase 1/2 | NCT03340974 | |
| Squamous cell carcinoma | Phase 1 | NCT01921426 | |
| Head and neck cancer | Phase 2 | NCT02508389 | |
| Metalloporphyrins | Lung cancer | Phase 3 | NCT00054795 |
| NOX inhibitors | |||
| Ebselen (SPI-1005) | Cancer | Phase 1 | NCT01452607 |
| Lung cancer, Head and neck cancer | Phase 2 | NCT01451853 | |
| GSH-related antioxidants | |||
| NAC | Breast cancer | Phase 1 | NCT01878695 |
| Gastric cancer | NA | NCT03238404 | |
| Ovarian cancer | NA | NCT03491033 | |
| Head and neck cancer | Phase 2 | NCT02123511 | |
| Gastrointestinal neoplasms | Phase 2 | NCT00196885 | |
| Bladder cancer | NA | NCT02756637 | |
| Lung cancer | Phase 2 | NCT00691132 | |
| Colorectal cancer | NA | NCT01325909 | |
| NOV-002 | Breast cancer | Phase 2 | NCT00499122 |
| Ovarian cancer | Phase 2 | NCT00345540 | |
| NSCLC | Phase 3 | NCT00347412 | |
| Leukemia | Phase 2 | NCT00960726 | |
| Reduced GSH | Breast cancer | Phase 2 | NCT00266331 |
| Vitamins | |||
| Vitamin C | Ovarian cancer | Phase 2 | NCT00284427 |
| Pancreatic cancer | Phase 1 | NCT00954525 | |
| Prostatic neoplasms | Phase 2 | NCT01080352 | |
| Ovarian cancer | Phase 2 | NCT00284427 | |
| Advanced cancer | Phase 1/2 | NCT01050621 | |
| Solid cancers | Phase 1 | NCT00441207 | |
| NSCLC | Phase 1/2 | NCT02655913 | |
| Head and Neck Cancer | NA | NCT03531190 | |
| Skin cancer | NA | NCT01032031 | |
| Liver cancer | Phase 1/2 | NCT01754987 | |
| Vitamin E | Prostate cancer | Phase 3 | NCT00006392 |
| Colorectal cancer | Phase 1 | NCT00905918 | |
| Head and neck neoplasms | Phase 2 | NCT02397486 | |
| Skin neoplasms | NA | NCT02248584 | |
| Pancreatic neoplasms | Phase 1 | NCT00985777 | |
| Breast cancer | Phase 2 | NCT00022204 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. https://doi.org/10.3390/antiox11061128
Luo M, Zhou L, Huang Z, Li B, Nice EC, Xu J, Huang C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants. 2022; 11(6):1128. https://doi.org/10.3390/antiox11061128
Chicago/Turabian StyleLuo, Maochao, Li Zhou, Zhao Huang, Bowen Li, Edouard C. Nice, Jia Xu, and Canhua Huang. 2022. "Antioxidant Therapy in Cancer: Rationale and Progress" Antioxidants 11, no. 6: 1128. https://doi.org/10.3390/antiox11061128
APA StyleLuo, M., Zhou, L., Huang, Z., Li, B., Nice, E. C., Xu, J., & Huang, C. (2022). Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants, 11(6), 1128. https://doi.org/10.3390/antiox11061128