
Chronic Intermittent Hypoxia-Induced Dysmetabolism Is Associated with Hepatic Oxidative Stress, Mitochondrial Dysfunction and Inflammation


 ,
,  , ,
, ,  , ,
, ,  ,
,  , and
, and 
Abstract
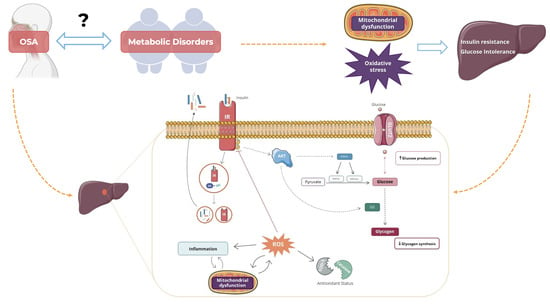
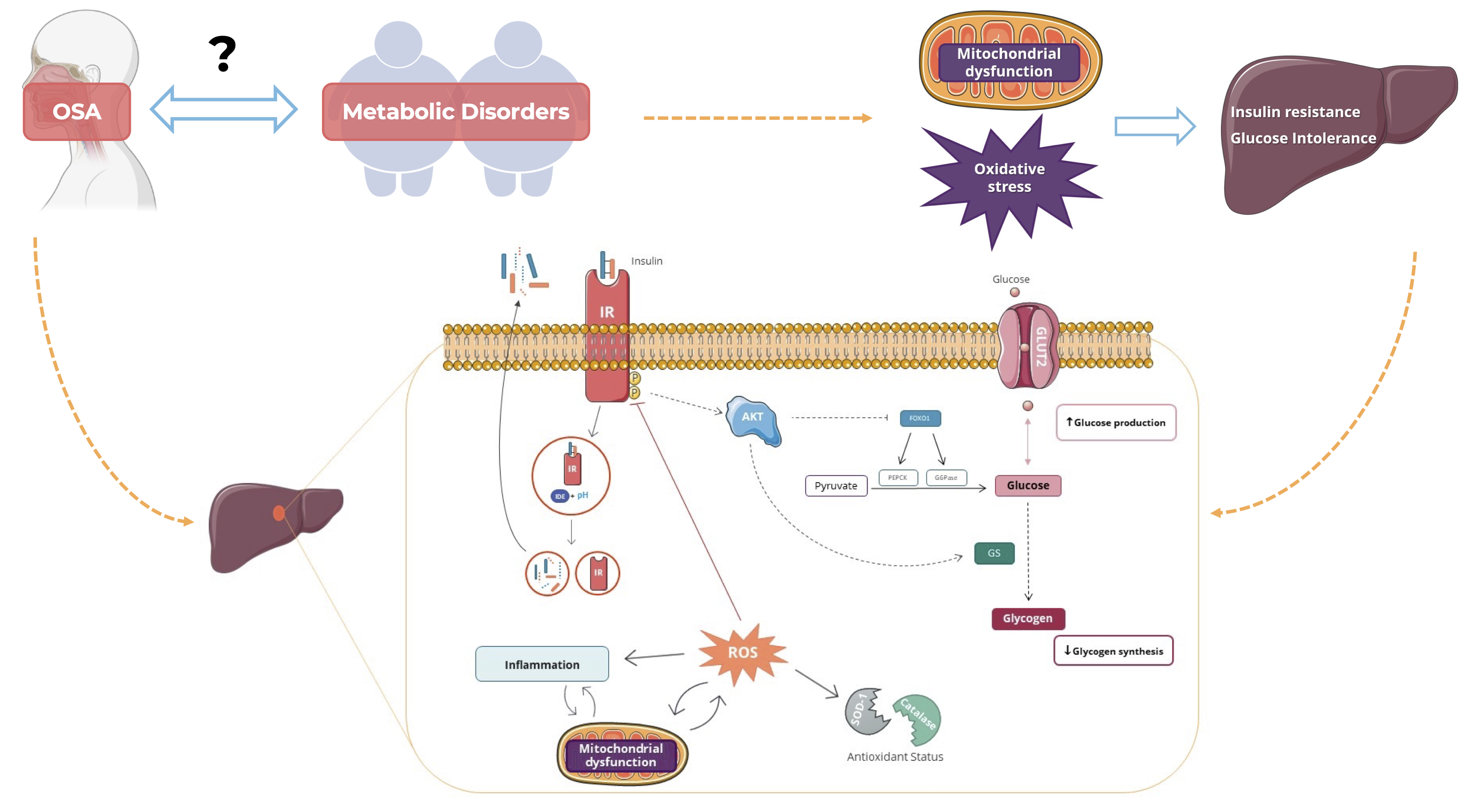
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and In Vivo Procedures
2.2. Assessment of the Levels of Key Proteins Involved in Insulin Signaling, Glucose Metabolism, Mitochondrial Biogenesis, Antioxidant System, and Inflammation Pathways
2.3. Histological Analysis of Lipid Deposition and Immunohistochemistry Analysis of Hypoxia Markers, Mitochondrial Density, and Inflammation in the Liver Tissue
2.4. Assessment of Mitochondrial Complexes’ Enzymatic Activity
2.5. Determination of Intracellular ROS Content, Lipid Peroxidation, and Antioxidant Capacity of Liver Tissue
2.6. Statistical Analysis
3. Results
3.1. Impact of Chronic Intermittent Hypoxia on Metabolic Parameters
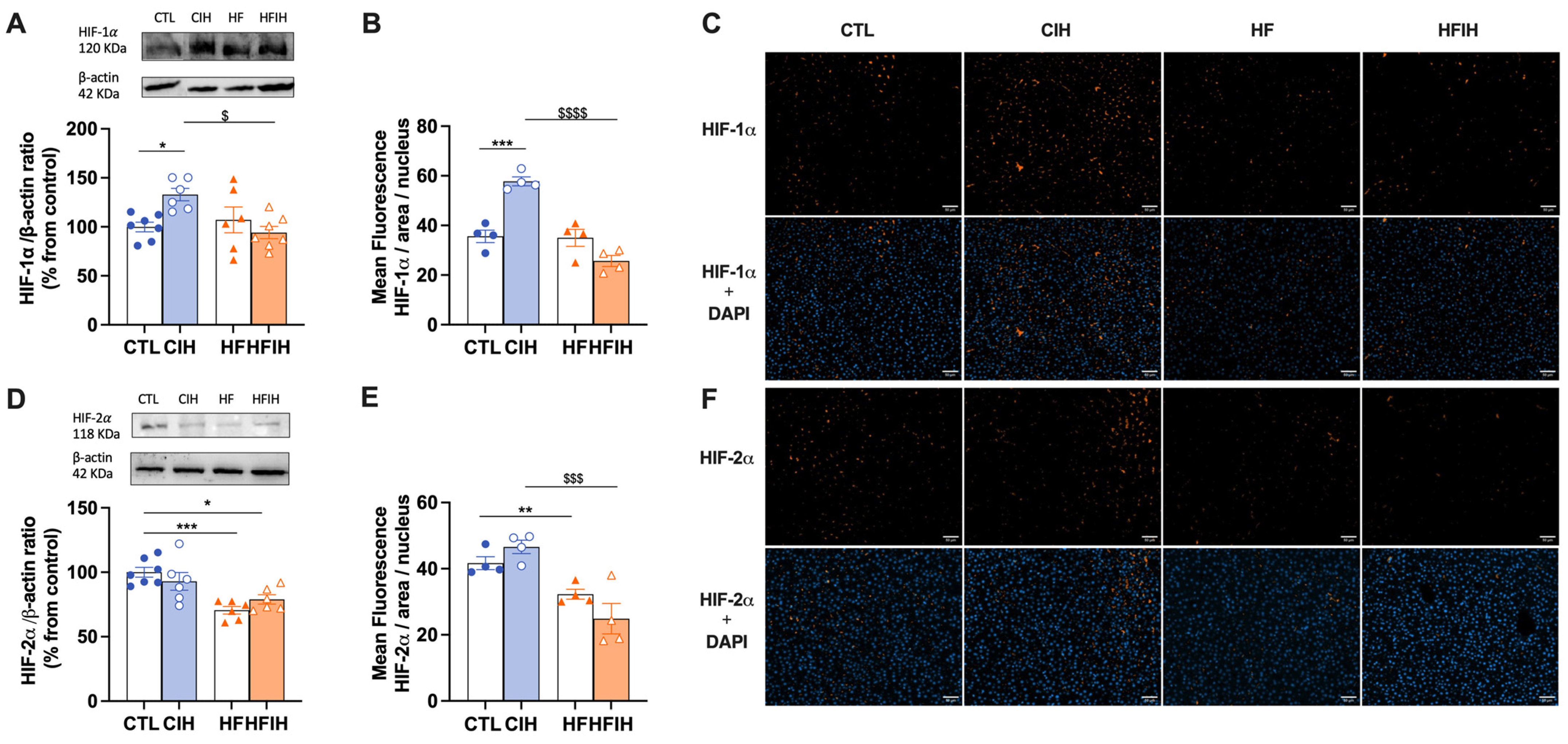
3.2. Effects of Chronic Intermittent Hypoxia on Hypoxia Markers
3.3. Effects of Chronic Intermittent Hypoxia on the Levels of Proteins Involved in Insulin Signalling and Glucose Uptake in the Liver
3.4. Effects of Chronic Intermittent Hypoxia on Mitochondrial Biogenesis and Activity
3.5. Effects of Chronic Intermittent Hypoxia on ROS Levels and on Antioxidant Capacity
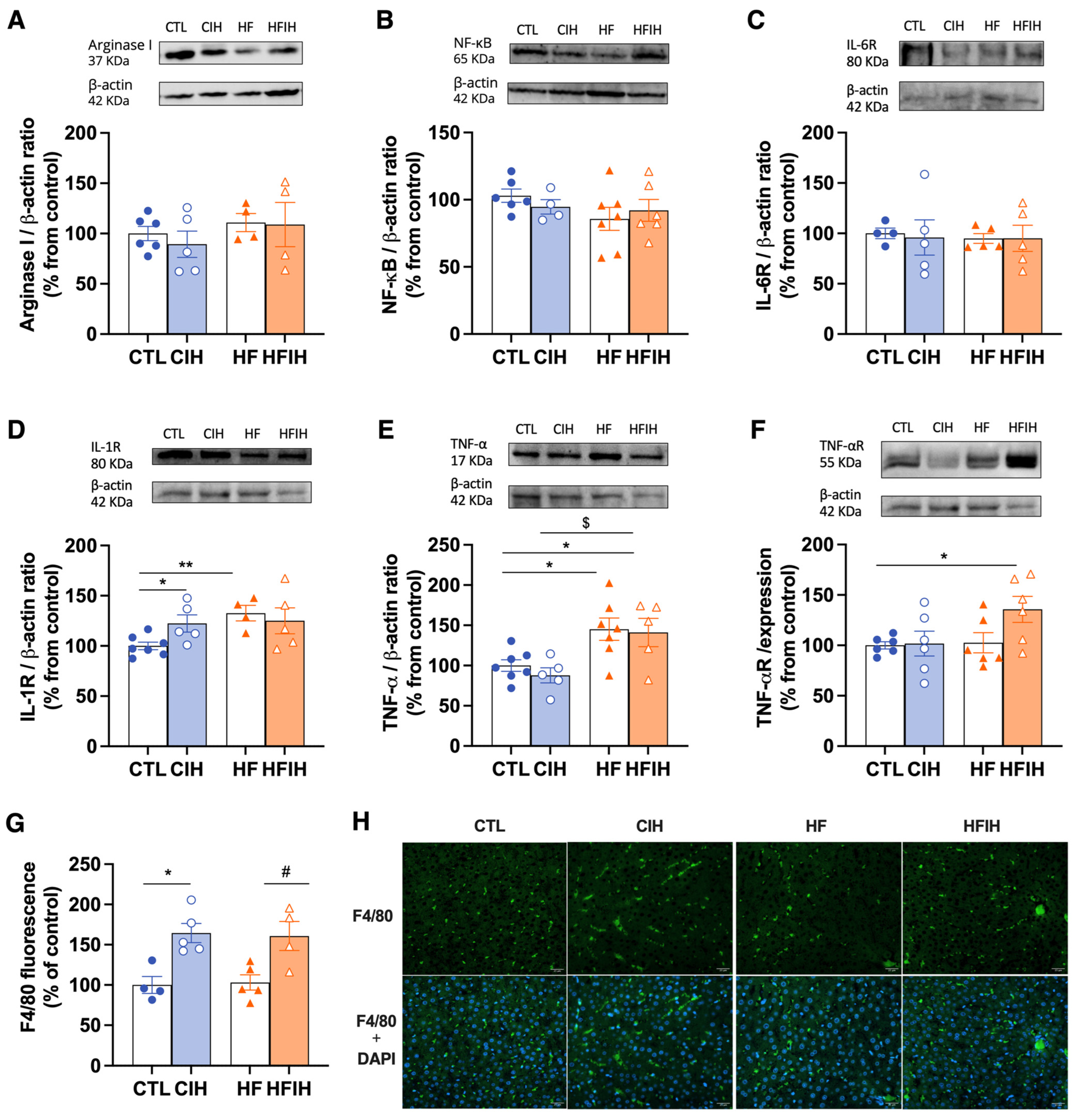
3.6. Effects of Chronic Intermittent Hypoxia on Inflammatory Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sateia, M.J. International Classification of Sleep Disorders—Third Edition Highlights and Modifications. Chest 2014, 146, 1387–1394. [Google Scholar] [CrossRef]
- Adir, Y.; Humbert, M.; Chaouat, A. Sleep-Related Breathing Disorders and Pulmonary Hypertension. Eur. Respir. J. 2021, 57, 2002258. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.S.; McSharry, D.G.; Malhotra, A. Adult Obstructive Sleep Apnoea. Lancet 2014, 383, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pépin, J.L. Obstructive Sleep Apnoea Syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef]
- Almendros, I.; Basoglu, Ö.K.; Conde, S.V.; Liguori, C.; Saaresranta, T. Metabolic Dysfunction in OSA: Is There Something New under the Sun? J. Sleep Res. 2022, 31, e13418. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, X.; Lu, Y. Obstructive Sleep Apnea Syndrome and Metabolic Diseases. Endocrinology 2018, 159, 2670–2675. [Google Scholar] [CrossRef]
- Bonsignore, M.R.; Borel, A.L.; Machan, E.; Grunstein, R. Sleep Apnoea and Metabolic Dysfunction. Eur. Respir. Rev. 2013, 22, 353–364. [Google Scholar] [CrossRef]
- Reutrakul, S.; Mokhlesi, B. Obstructive Sleep Apnea and Diabetes: A State of the Art Review. Chest 2017, 152, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Conde, S.V.; Sacramento, J.F.; Guarino, M.P.; Gonzalez, C.; Obeso, A.; Diogo, L.N.; Monteiro, E.C.; Ribeiro, M.J. Carotid Body, Insulin and Metabolic Diseases: Unravelling the Links. Front. Physiol. 2014, 5, 418. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The Prevalence and Incidence of NAFLD Worldwide: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical Epidemiology and Disease Burden of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 8263. [Google Scholar] [CrossRef]
- Parikh, M.P.; Gupta, N.M.; McCullough, A.J. Obstructive Sleep Apnea and the Liver. Clin. Liver Dis. 2019, 23, 363–382. [Google Scholar] [CrossRef]
- Ji, Y.; Liang, Y.; Mak, J.C.W.; Ip, M.S.M. Obstructive Sleep Apnea, Intermittent Hypoxia and Non-Alcoholic Fatty Liver Disease. Sleep Med. 2022, 95, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Mesarwi, O.A.; Loomba, R.; Malhotra, A. Obstructive Sleep Apnea, Hypoxia, and Nonalcoholic Fatty Liver Disease. Am. J. Respir. Crit. Care Med. 2019, 199, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.O.; Sacramento, J.F.; Olea, E.; Melo, B.F.; Prieto-Lloret, J.; Obeso, A.; Rocher, A.; Matafome, P.; Monteiro, E.C.; Conde, S.V. Chronic Intermittent Hypoxia Induces Early-Stage Metabolic Dysfunction Independently of Adipose Tissue Deregulation. Antioxidants 2021, 10, 1233. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martín, M.C.; Vega-Agapito, M.V.; Conde, S.V.; Castañeda, J.; Bustamante, R.; Olea, E.; Perez-Vizcaino, F.; Gonzalez, C.; Obeso, A. Carotid Body Function and Ventilatory Responses in Intermittent Hypoxia. Evidence for Anomalous Brainstem Integration of Arterial Chemoreceptor Input. J. Cell. Physiol. 2011, 226, 1961–1969. [Google Scholar] [CrossRef]
- Olea, E.; Agapito, M.T.; Gallego-Martin, T.; Rocher, A.; Gomez-Niño, A.; Obeso, A.; Gonzalez, C.; Yubero, S. Intermittent Hypoxia and Diet-Induced Obesity: Effects on Oxidative Status, Sympathetic Tone, Plasma Glucose and Insulin Levels, and Arterial Pressure. J. Appl. Physiololy 2014, 117, 706–719. [Google Scholar] [CrossRef]
- Conde, S.V.; Ribeiro, M.J.; Obeso, A.; Rigual, R.; Monteiro, E.C.; Gonzalez, C. Chronic Caffeine Intake in Adult Rat Inhibits Carotid Body Sensitization Produced by Chronic Sustained Hypoxia but Maintains Intact Chemoreflex Output. Mol. Pharmacol. 2012, 82, 1056–1065. [Google Scholar] [CrossRef]
- Fiji App for ImageJ. Available online: https://imagej.net/software/fiji/ (accessed on 13 October 2023).
- Crisostomo, L.; Videira, R.A.; Jarak, I.; Starčević, K.; Mašek, T.; Rato, L.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Diet during Early Life Defines Testicular Lipid Content and Sperm Quality in Adulthood. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1061–E1073. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.J.; Pimpão, A.B.; Lopes-Coelho, F.; Sequeira, C.O.; Coelho, N.R.; Gonçalves-Dias, C.; Barouki, R.; Coumoul, X.; Serpa, J.; Morello, J.; et al. Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)Adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex. Antioxidants 2021, 10, 1484. [Google Scholar] [CrossRef] [PubMed]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinolology Metab. 2017, 28, 497. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Al-Hasani, H. Glucose Transporters in Adipose Tissue, Liver, and Skeletal Muscle in Metabolic Health and Disease. Pflügers Arch.—Eur. J. Physiol. 2020, 472, 1273. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and Maladaptive Cardiorespiratory Responses to Continuous and Intermittent Hypoxia Mediated by Hypoxia-Inducible Factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Punjabi, N.M.; Beamer, B.A. Alterations in Glucose Disposal in Sleep-Disordered Breathing. Am. J. Respir. Crit. Care Med. 2012, 179, 235–240. [Google Scholar] [CrossRef]
- Louis, M.; Punjabi, N.M. Effects of Acute Intermittent Hypoxia on Glucose Metabolism in Awake Healthy Volunteers. J. Appl. Physiol. (1985) 2009, 106, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2012, 2, 1117–1133. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Q.; Wu, X.; Zhang, Y.; Xing, X.; Lin, K.; Feng, Y.; Wang, M.; Wang, Y.; Wang, R. Hypoxia as a Double-Edged Sword to Combat Obesity and Comorbidities. Cells 2022, 11, 3735. [Google Scholar] [CrossRef]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef]
- Sacramento, J.F.; Ribeiro, M.J.; Rodrigues, T.; Guarino, M.P.; Diogo, L.N.; Seiça, R.; Monteiro, E.C.; Matafome, P.; Conde, S.V. Insulin Resistance Is Associated with Tissue-Specific Regulation of HIF-1α and HIF-2α during Mild Chronic Intermittent Hypoxia. Respir. Physiol. Neurobiol. 2016, 228, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.; Carvou, N.J.C.; Debnam, E.S.; Srai, S.K.; Unwin, R.J. Diabetes Increases Facilitative Glucose Uptake and GLUT2 Expression at the Rat Proximal Tubule Brush Border Membrane. J. Physiol. 2003, 553, 137–145. [Google Scholar] [CrossRef]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The Aetiology and Molecular Landscape of Insulin Resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Minville, C.; Tordjman, J.; Lévy, P.; Bouillot, J.L.; Basdevant, A.; Bedossa, P.; Clément, K.; Pépin, J.L. Chronic Intermittent Hypoxia Is a Major Trigger for Non-Alcoholic Fatty Liver Disease in Morbid Obese. J. Hepatol. 2012, 56, 225–233. [Google Scholar] [CrossRef]
- Li, J.; Thorne, L.N.; Punjabi, N.M.; Sun, C.K.; Schwartz, A.R.; Smith, P.L.; Marino, R.L.; Rodriguez, A.; Hubbard, W.C.; O’Donnell, C.P.; et al. Intermittent Hypoxia Induces Hyperlipidemia in Lean Mice. Circ. Res. 2005, 97, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, D.O.; Khita, O.O.; Tsymbal, D.O.; Danilovskyi, S.V.; Rudnytska, O.V.; Halkin, O.V.; Kryvdiuk, I.V.; Smeshkova, M.V.; Yakymchuk, M.M.; Bezrodnyi, B.H.; et al. Expression of IDE and PITRM1 Genes in ERN1 Knockdown U87 Glioma Cells: Effect of Hypoxia and Glucose Deprivation. Endocr. Regul. 2020, 54, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-Inducible Factor 2 Regulates Hepatic Lipid Metabolism. Mol. Cell. Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Cheng, K.; Ho, K.; Stokes, R.; Scott, C.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Loudovaris, T.; Kay, T.W.; Kulkarni, R.N.; et al. Hypoxia-Inducible Factor-1α Regulates β Cell Function in Mouse and Human Islets. J. Clin. Investig. 2010, 120, 2171–2183. [Google Scholar] [CrossRef]
- Catrina, S.B.; Zheng, X. Hypoxia and Hypoxia-Inducible Factors in Diabetes and Its Complications. Diabetologia 2021, 64, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Gonzalez-Martin, M.D.C.; Vega-Agapito, V.; Gonzalez, C.; Obeso, A.; Farré, R.; Agapito, T.; Yubero, S. The Effects of Intermittent Hypoxia on Redox Status, NF-ΚB Activation, and Plasma Lipid Levels Are Dependent on the Lowest Oxygen Saturation. Free. Radic. Biol. Med. 2013, 65, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.M.; Liu, J.; Theodore, J.; Robin, E.D. Effect of Hyperoxia, Hypoxia, and Maturation on Superoxide Dismutase Activity in Isolated Alveolar Macrophages. Am. Rev. Respir. Dis. 1977, 115, 279–284. [Google Scholar]
- Morita, M.; Ishida, N.; Uchiyama, K.; Yamaguchi, K.; Itoh, Y.; Shichiri, M.; Yoshida, Y.; Hagihara, Y.; Naito, Y.; Yoshikawa, T.; et al. Fatty Liver Induced by Free Radicals and Lipid Peroxidation. Free Radic. Res. 2012, 46, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.J.; Pimpão, A.B.; Fernandes, D.G.F.; Morello, J.; Sequeira, C.O.; Calado, J.; Antunes, A.M.M.; Almeida, M.S.; Branco, P.; Monteiro, E.C.; et al. Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine. Molecules 2022, 27, 1416. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.C.; Ramos, C.; Santos, I.; Mendes, C.; Silva, F.; Vicente, J.B.; Pereira, S.A.; Félix, A.; Gonçalves, L.G.; Serpa, J. Cysteine Boosts Fitness Under Hypoxia-Mimicked Conditions in Ovarian Cancer by Metabolic Reprogramming. Front. Cell Dev. Biol. 2021, 9, 2107. [Google Scholar] [CrossRef]
- Chen, Q.; Han, X.; Chen, M.; Zhao, B.; Sun, B.; Sun, L.; Zhang, W.; Yu, L.; Liu, Y. High-Fat Diet-Induced Mitochondrial Dysfunction Promotes Genioglossus Injury—A Potential Mechanism for Obstructive Sleep Apnea with Obesity. Nat. Sci. Sleep 2021, 13, 2203–2219. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 Controls Mitochondrial Metabolism during Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α Protects against Oxidative Stress by Directly Targeting Mitochondria. Redox Biol. 2019, 25, 101119. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. Innov. Hepatol. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Cornejo, J.C.; Polakos, N.K.; John, B.; Wuensch, S.A.; Topham, D.J.; Pierce, R.H.; Crispe, I.N. Kupffer cell heterogeneity: Functional properties of bone marrow derived and sessile hepatic macrophages. Blood 2007, 110, 4077–4085. [Google Scholar] [CrossRef]
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [Google Scholar] [CrossRef]
- Xu, G.X.; Wei, S.; Yu, C.; Zhao, S.Q.; Yang, W.J.; Feng, Y.H.; Pan, C.; Yang, K.X.; Ma, Y. Activation of Kupffer cells in NAFLD and NASH: Mechanisms and therapeutic interventions. Front. Cell Dev. Biol. 2023, 11, 1199519. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, J.L.; Martins, F.O.; Olea, E.; Prieto-Lloret, J.; Braga, P.C.; Sacramento, J.F.; Sequeira, C.O.; Negrinho, A.P.; Pereira, S.A.; Alves, M.G.; et al. Chronic Intermittent Hypoxia-Induced Dysmetabolism Is Associated with Hepatic Oxidative Stress, Mitochondrial Dysfunction and Inflammation. Antioxidants 2023, 12, 1910. https://doi.org/10.3390/antiox12111910
Fernandes JL, Martins FO, Olea E, Prieto-Lloret J, Braga PC, Sacramento JF, Sequeira CO, Negrinho AP, Pereira SA, Alves MG, et al. Chronic Intermittent Hypoxia-Induced Dysmetabolism Is Associated with Hepatic Oxidative Stress, Mitochondrial Dysfunction and Inflammation. Antioxidants. 2023; 12(11):1910. https://doi.org/10.3390/antiox12111910
Chicago/Turabian StyleFernandes, Joana L., Fátima O. Martins, Elena Olea, Jesus Prieto-Lloret, Patrícia C. Braga, Joana F. Sacramento, Catarina O. Sequeira, Ana P. Negrinho, Sofia A. Pereira, Marco G. Alves, and et al. 2023. "Chronic Intermittent Hypoxia-Induced Dysmetabolism Is Associated with Hepatic Oxidative Stress, Mitochondrial Dysfunction and Inflammation" Antioxidants 12, no. 11: 1910. https://doi.org/10.3390/antiox12111910
APA StyleFernandes, J. L., Martins, F. O., Olea, E., Prieto-Lloret, J., Braga, P. C., Sacramento, J. F., Sequeira, C. O., Negrinho, A. P., Pereira, S. A., Alves, M. G., Rocher, A., & Conde, S. V. (2023). Chronic Intermittent Hypoxia-Induced Dysmetabolism Is Associated with Hepatic Oxidative Stress, Mitochondrial Dysfunction and Inflammation. Antioxidants, 12(11), 1910. https://doi.org/10.3390/antiox12111910