

Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatments
2.3. Animal Model and Treatment Strategies
2.4. Levels of Serum Creatinine, Blood Urea Nitrogen (BUN), and Indoxyl Sulfate
2.5. Histological Analysis
2.6. Immunohistochemistry (IHC) Staining
2.7. Iron Stain
2.8. Immunoblotting and Antibodies
2.9. MTT Assay
2.10. DCFDA Cellular ROS Detection Assay
2.11. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
2.12. Measurement of Cellular Iron Levels
2.13. Detection of Fe2+ Using Fluorescent Probes
2.14. Lipid Peroxidation Assay
2.15. Total and Reduced Glutathione (GSH) Measurement
2.16. Statistical Analysis
3. Results
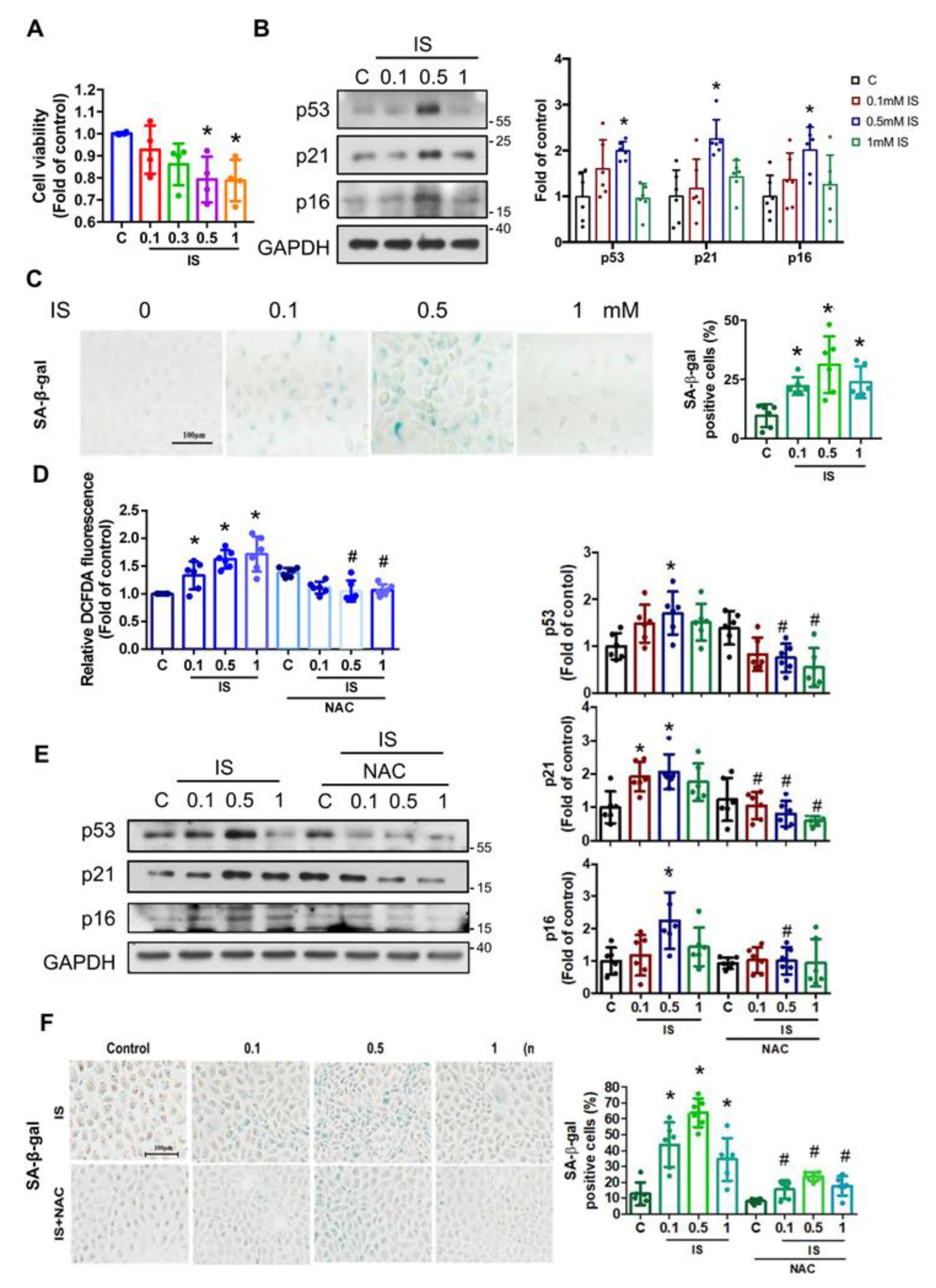
3.1. Indoxyl Sulfate (IS) Induces Senescence through ROS in Renal Tubular Cells
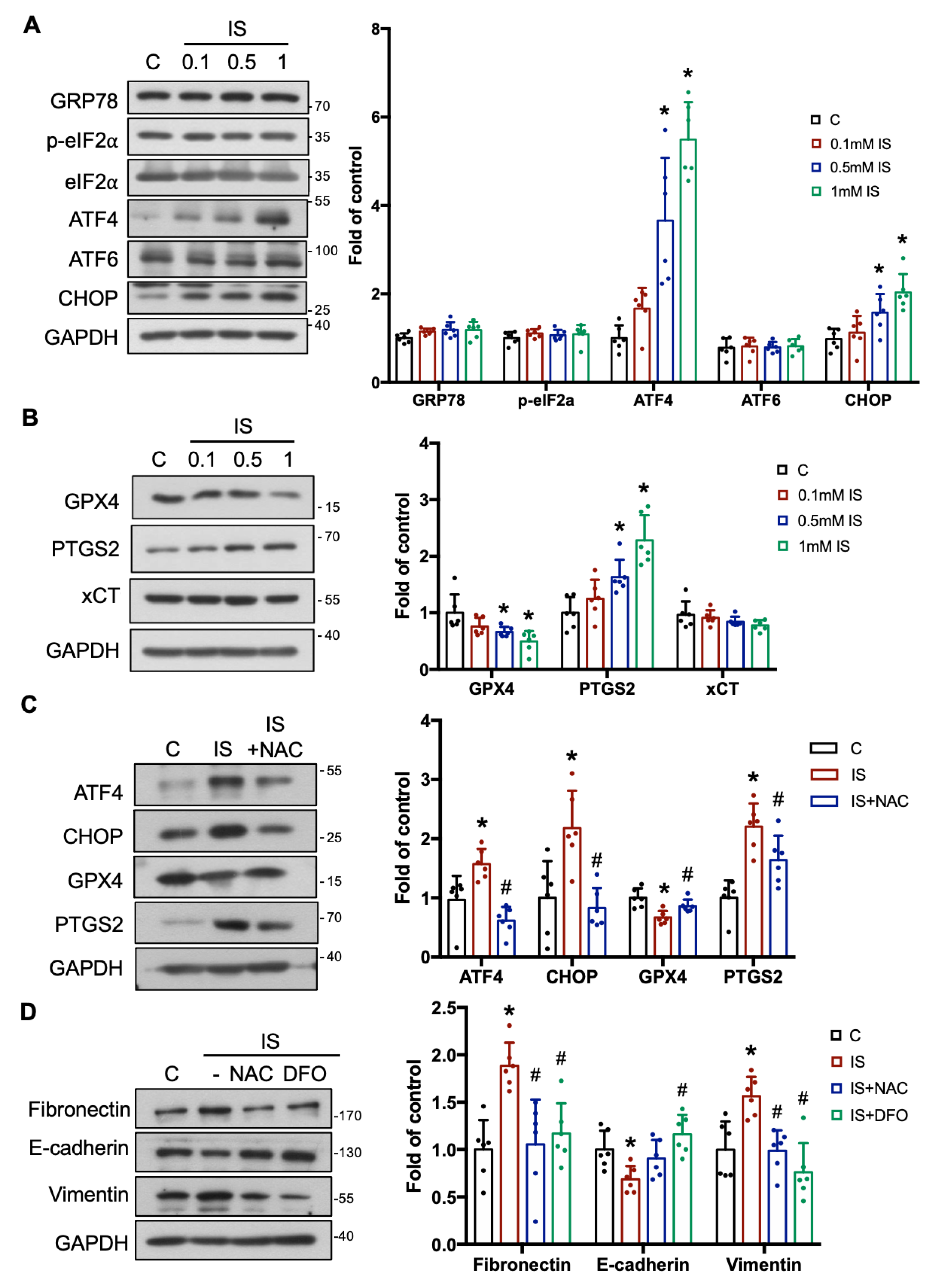
3.2. IS Triggers Renal Fibrosis/Injury via ER Stress, Ferroptosis, and Epithelial–Mesenchymal Transition (EMT)
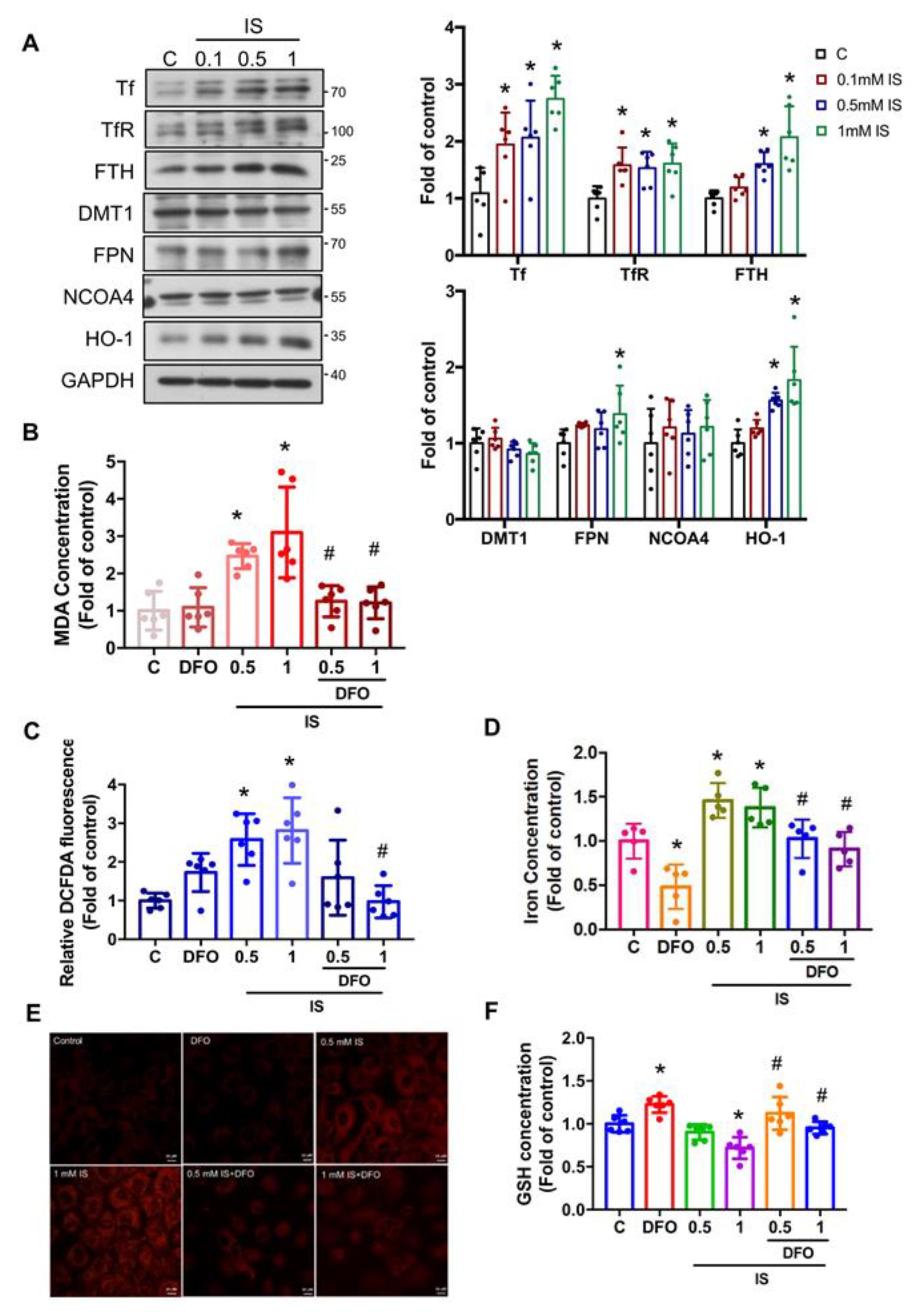
3.3. IS Induces the Accumulation of Intracellular Iron and Causes Ferroptosis in Renal Tubular Cells
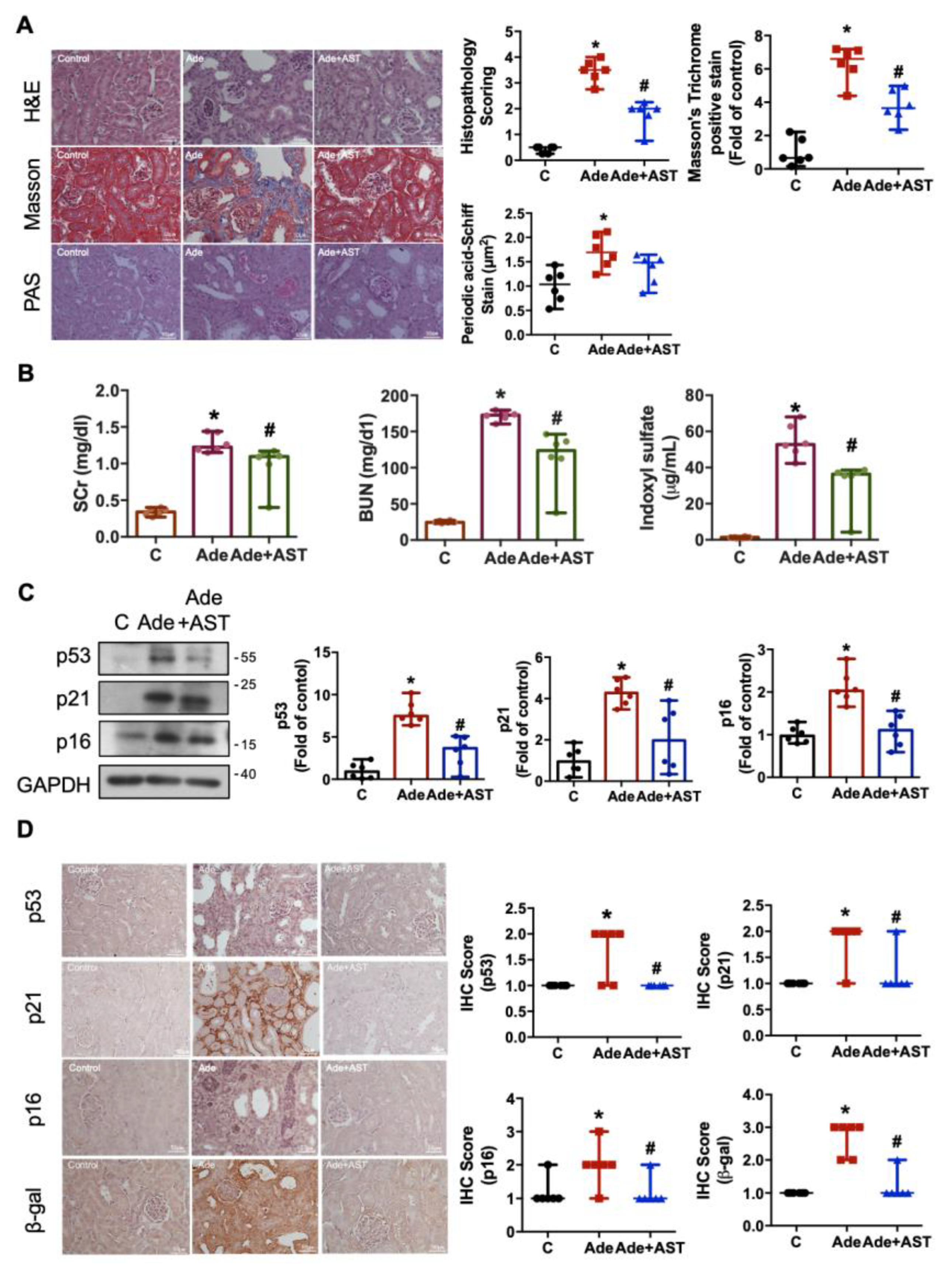
3.4. IS Accumulation Causes Renal Injury and Senescence in Adenine-Induced CKD Mice
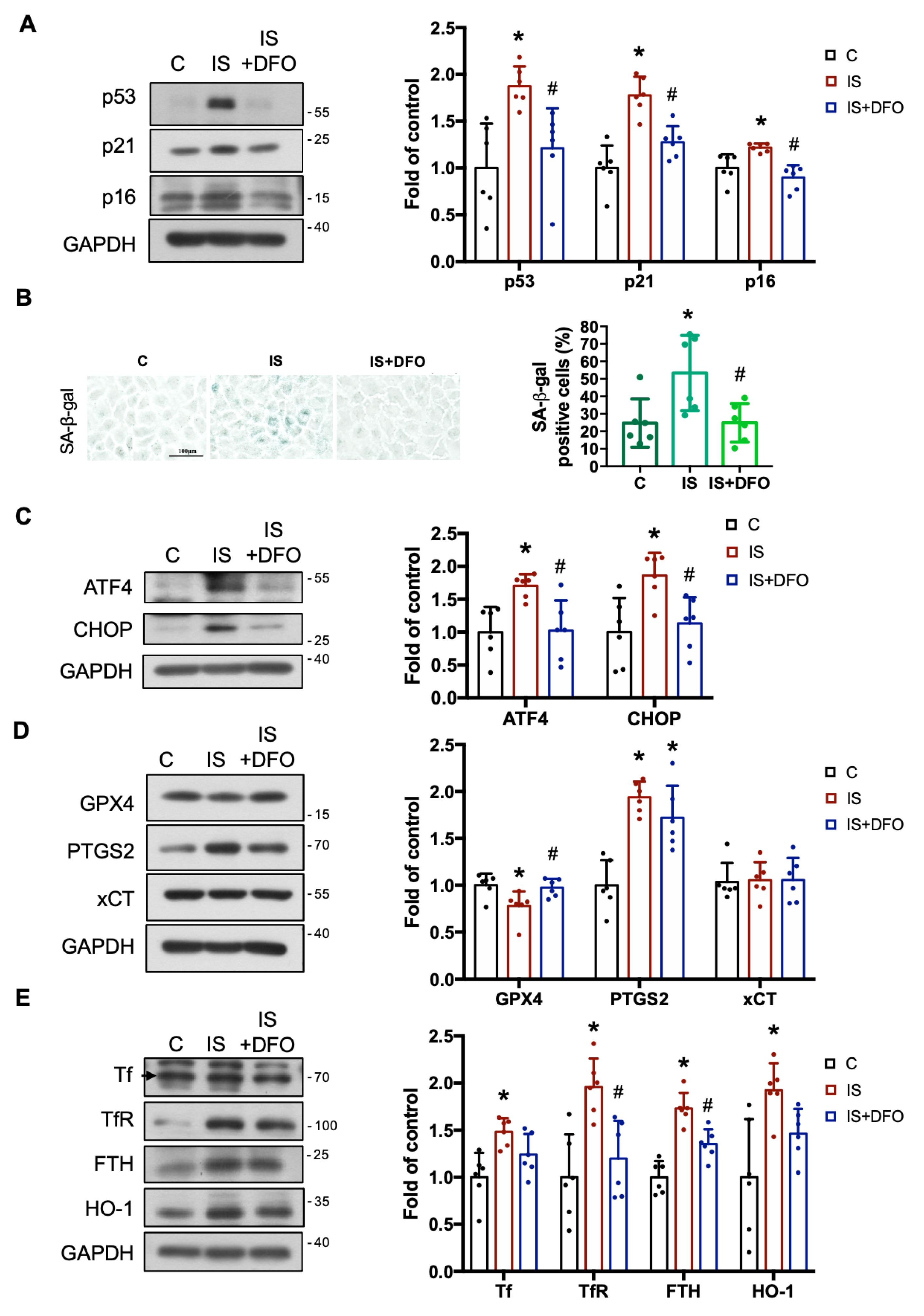
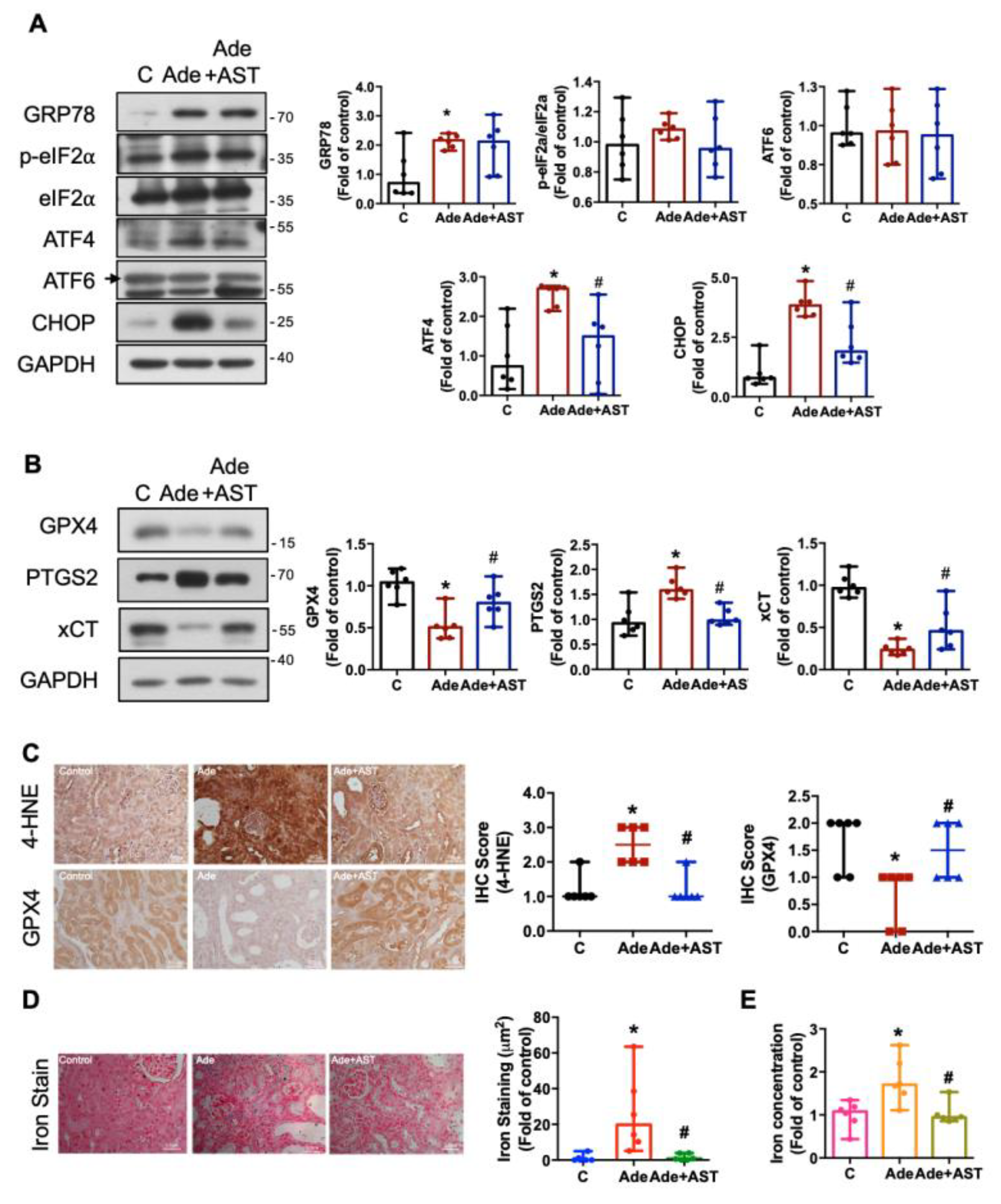
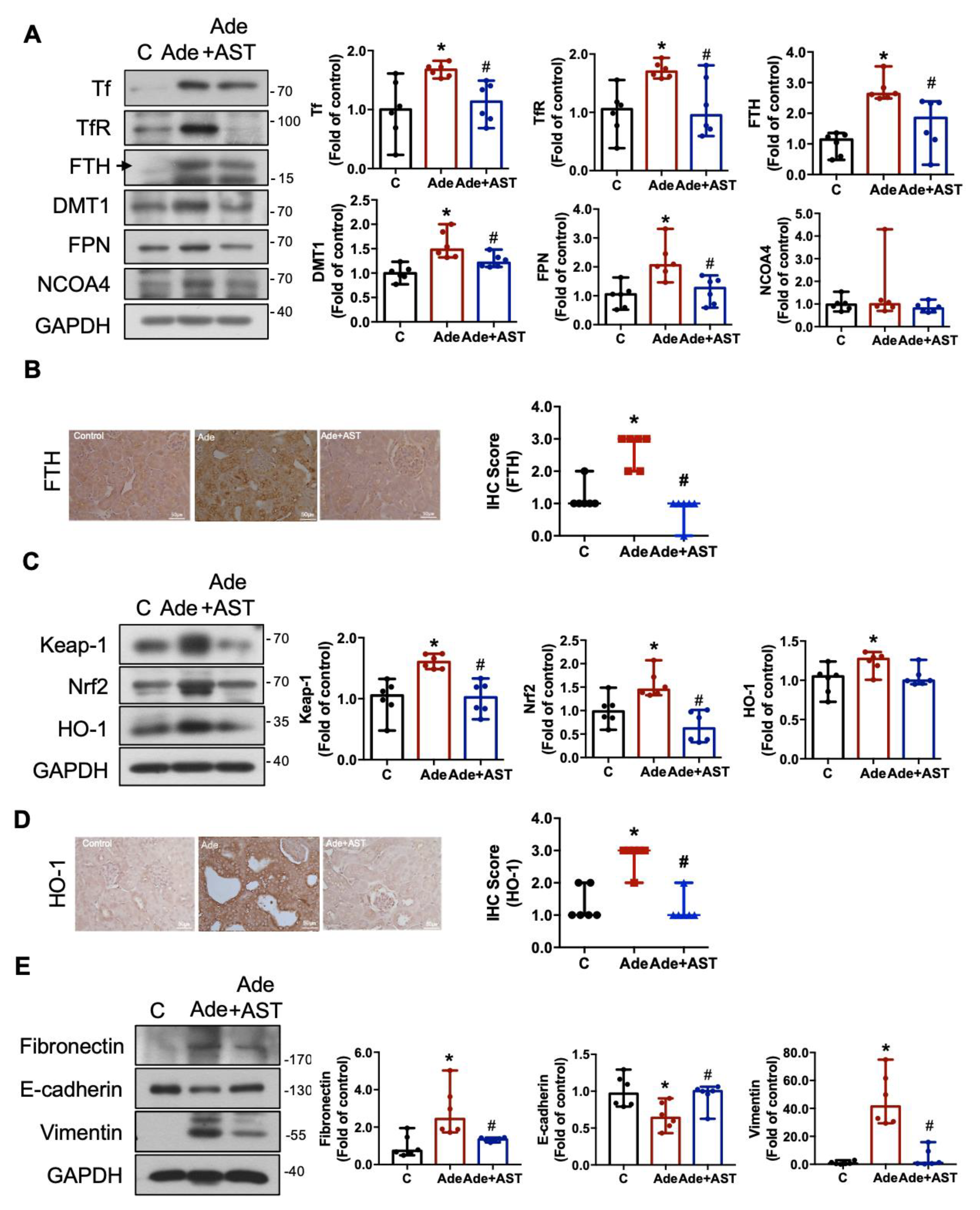
3.5. IS Contributes to ER Stress, Ferroptosis, Iron Accumulation, Nrf2/HO-1 Activation, and the EMT Process in CKD Kidneys
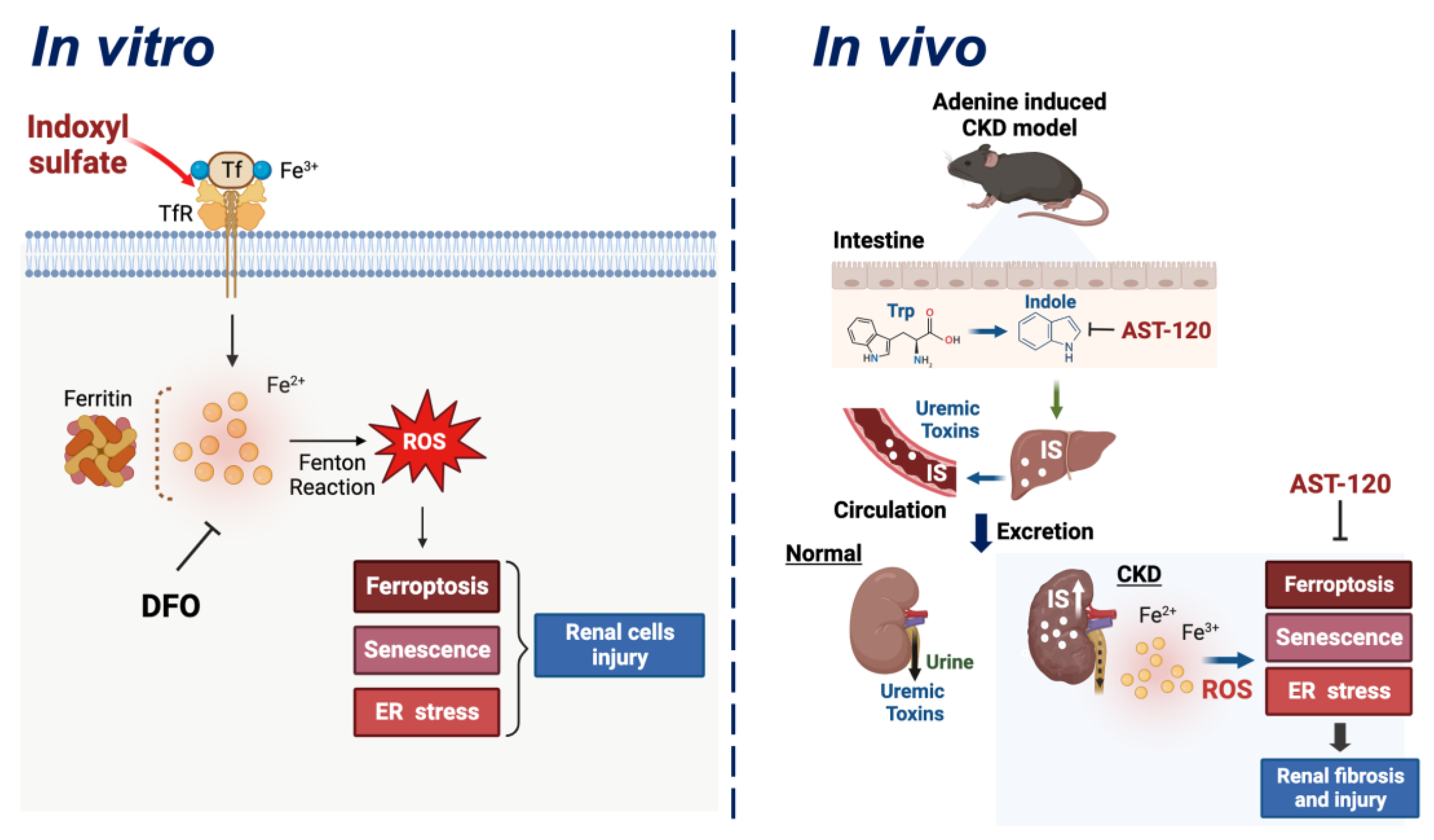
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallee, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef]
- Wakabayashi, I.; Marumo, M. Evidence for Indoxyl Sulfate as an Inducer of Oxidative Stress in Patients with Diabetes. In Vivo 2022, 36, 1790–1794. [Google Scholar] [CrossRef]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Zununi Vahed, S.; Mostafavi, S.; Hosseiniyan Khatibi, S.M.; Shoja, M.M.; Ardalan, M. Vascular Calcification: An Important Understanding in Nephrology. Vasc. Health Risk Manag. 2020, 16, 167–180. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Vida, C.; Bodega, G.; Ceprian, N.; Morales, E.; Praga, M.; de Sequera, P.; Ramirez, R. Mechanisms of Cardiovascular Disorders in Patients with Chronic Kidney Disease: A Process Related to Accelerated Senescence. Front. Cell Dev. Biol. 2020, 8, 185. [Google Scholar] [CrossRef]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef]
- Kawakami, T.; Inagi, R.; Wada, T.; Tanaka, T.; Fujita, T.; Nangaku, M. Indoxyl sulfate inhibits proliferation of human proximal tubular cells via endoplasmic reticulum stress. Am. J. Physiol. Ren. Physiol. 2010, 299, F568–F576. [Google Scholar] [CrossRef]
- Diwan, V.; Brown, L.; Gobe, G.C. Adenine-induced chronic kidney disease in rats. Nephrology 2018, 23, 5–11. [Google Scholar] [CrossRef]
- Yokozawa, T.; Zheng, P.D.; Oura, H.; Koizumi, F. Animal model of adenine-induced chronic renal failure in rats. Nephron 1986, 44, 230–234. [Google Scholar] [CrossRef]
- Rahman, A.; Yamazaki, D.; Sufiun, A.; Kitada, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. A novel approach to adenine-induced chronic kidney disease associated anemia in rodents. PLoS ONE 2018, 13, e0192531. [Google Scholar] [CrossRef]
- Cheng, T.H.; Ma, M.C.; Liao, M.T.; Zheng, C.M.; Lu, K.C.; Liao, C.H.; Hou, Y.C.; Liu, W.C.; Lu, C.L. Indoxyl Sulfate, a Tubular Toxin, Contributes to the Development of Chronic Kidney Disease. Toxins 2020, 12, 684. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Yamamoto, H.; Okamoto, A.; Imanishi, K.; Tokui, N.; Okamoto, T.; Suzuki, Y.; Sugiyama, N.; Imai, A.; Kudo, S.; et al. Effect of an Oral Adsorbent, AST-120, on Dialysis Initiation and Survival in Patients with Chronic Kidney Disease. Int. J. Nephrol. 2012, 2012, 376128. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Zhao, G.; Arosio, P.; Chasteen, N.D. Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry 2006, 45, 3429–3436. [Google Scholar] [CrossRef]
- Naito, Y.; Fujii, A.; Sawada, H.; Oboshi, M.; Iwasaku, T.; Okuhara, Y.; Morisawa, D.; Eguchi, A.; Hirotani, S.; Masuyama, T. Association between renal iron accumulation and renal interstitial fibrosis in a rat model of chronic kidney disease. Hypertens. Res. 2015, 38, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Wang, C.C.; Loh, J.Z.; Chiang, T.C.; Weng, T.I.; Chan, D.C.; Hung, K.Y.; Chiang, C.K.; Liu, S.H. Therapeutic Ultrasound Halts Progression of Chronic Kidney Disease In Vivo via the Regulation of Markers Associated with Renal Epithelial-Mesenchymal Transition and Senescence. Int. J. Mol. Sci. 2022, 23, 13387. [Google Scholar] [CrossRef]
- Tan, X.; Cao, X.S.; Zhang, P.; Xiang, F.F.; Teng, J.; Zou, J.Z.; Ding, X.Q. Endoplasmic reticulum stress associated apoptosis as a novel mechanism in indoxyl sulfate-induced cardiomyocyte toxicity. Mol. Med. Rep. 2018, 18, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yu, D.; He, Z.; Bao, L.; Feng, L.; Chen, L.; Liu, Z.; Hu, X.; Zhang, N.; Wang, T.; et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free. Radic. Biol. Med. 2021, 175, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Investig. 2012, 92, 488–498. [Google Scholar] [CrossRef]
- Chiang, C.K.; Hsu, S.P.; Wu, C.T.; Huang, J.W.; Cheng, H.T.; Chang, Y.W.; Hung, K.Y.; Wu, K.D.; Liu, S.H. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol. Med. 2011, 17, 1295–1305. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Li, Y.Y.; Zhang, X.Y.; Tian, Z.H.; Liu, C.; Wang, S.T.; Zhang, F.R. Cellular senescence of renal tubular epithelial cells in renal fibrosis. Front. Endocrinol. 2023, 14, 1085605. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Cheng, M.; Zhang, L.; Wang, Y.P. Ferroptosis and renal fibrosis: A new target for the future (Review). Exp. Ther. Med. 2023, 25, 13. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef]
- Tang, Z.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Zhang, D.; Gao, H.; Sun, H.; Zhang, J.; Gu, P. HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 2021, 43, 101971. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Leong, X.F. Lipid Oxidation Products on Inflammation-Mediated Hypertension and Atherosclerosis: A Mini Review. Front. Nutr. 2021, 8, 717740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Jing, S.; Lu, Y.; Zhang, J.; Ren, Y.; Mo, Y.; Liu, D.; Duan, L.; Yuan, Z.; Wang, C.; Wang, Q. Levistilide a Induces Ferroptosis by Activating the Nrf2/HO-1 Signaling Pathway in Breast Cancer Cells. Drug Des. Devel. Ther. 2022, 16, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Tomino, Y.; Lu, K.C. Impacts of Indoxyl Sulfate and p-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef]
- Nakano, T.; Watanabe, H.; Imafuku, T.; Tokumaru, K.; Fujita, I.; Arimura, N.; Maeda, H.; Tanaka, M.; Matsushita, K.; Fukagawa, M.; et al. Indoxyl Sulfate Contributes to mTORC1-Induced Renal Fibrosis via the OAT/NADPH Oxidase/ROS Pathway. Toxins 2021, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Henaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Nakagawa, K.; Itoya, M.; Takemoto, N.; Matsuura, Y.; Tawa, M.; Matsumura, Y.; Ohkita, M. Indoxyl sulfate induces ROS production via the aryl hydrocarbon receptor-NADPH oxidase pathway and inactivates NO in vascular tissues. Life Sci. 2021, 265, 118807. [Google Scholar] [CrossRef]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-kappaB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef]
- van Raaij, S.; van Swelm, R.; Bouman, K.; Cliteur, M.; van den Heuvel, M.C.; Pertijs, J.; Patel, D.; Bass, P.; van Goor, H.; Unwin, R.; et al. Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci. Rep. 2018, 8, 9353. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nishiya, K.; Ito, H.; Hosokawa, T.; Hashimoto, K.; Moriki, T. Iron deposition in renal biopsy specimens from patients with kidney diseases. Am. J. Kidney Dis. 2001, 38, 1038–1044. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Wu, X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. 2022, 13, 182. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Yu, X.; Qiao, Y.; Pan, S.; Wang, R.; Zheng, B.; Wang, H.; Ren, K.D.; Liu, H.; Yang, Y. Ferroptosis and Acute Kidney Injury (AKI): Molecular Mechanisms and Therapeutic Potentials. Front. Pharmacol. 2022, 13, 858676. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, D.H.; Choi, H.I.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. 3-Carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) induces cell death through ferroptosis and acts as a trigger of apoptosis in kidney cells. Cell Death Dis. 2023, 14, 78. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Gear, A.R. Observations on iron uptake, iron metabolism, cytochrome c content, cytochrome a content and cytochrome c-oxidase activity in regenerating rat liver. Biochem. J. 1965, 97, 532–539. [Google Scholar] [CrossRef]
- Martines, A.M.; Masereeuw, R.; Tjalsma, H.; Hoenderop, J.G.; Wetzels, J.F.; Swinkels, D.W. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nat. Rev. Nephrol. 2013, 9, 385–398. [Google Scholar] [CrossRef]
- Leaf, D.E.; Swinkels, D.W. Catalytic iron and acute kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 311, F871–F876. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Okar, L.; Iqbal, P.; Yassin, M.A. Iatrogenic Iron Overload in a Patient with Chronic Kidney Disease: Is There a Correlation Between Serum Ferritin and Liver Iron Concentration Determined by MRI T2*? Cureus 2020, 12, e8914. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Dixon, S.J.; Tyurina, Y.Y.; Kellum, J.A.; Kagan, V.E. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat. Rev. Nephrol. 2023, 19, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Holden, P.; Nair, L.S. Deferoxamine: An Angiogenic and Antioxidant Molecule for Tissue Regeneration. Tissue Eng. Part B Rev. 2019, 25, 461–470. [Google Scholar] [CrossRef]
- Ye, Y.; Chen, A.; Li, L.; Liang, Q.; Wang, S.; Dong, Q.; Fu, M.; Lan, Z.; Li, Y.; Liu, X.; et al. Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int. 2022, 102, 1259–1275. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ozono, I.; Tajima, S.; Imao, M.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; Tsuchiya, K.; et al. Iron chelation by deferoxamine prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. PLoS ONE 2014, 9, e89355. [Google Scholar] [CrossRef]
- Tan, T.C.; Crawford, D.H.; Jaskowski, L.A.; Subramaniam, V.N.; Clouston, A.D.; Crane, D.I.; Bridle, K.R.; Anderson, G.J.; Fletcher, L.M. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Lab. Investig. 2013, 93, 1295–1312. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Cha, H.J.; Choi, E.O.; Kim, C.H.; Kim, G.Y.; Yoo, Y.H.; Hwang, H.J.; Park, H.T.; Yoon, H.M.; Choi, Y.H. Activation of the Nrf2/HO-1 signaling pathway contributes to the protective effects of baicalein against oxidative stress-induced DNA damage and apoptosis in HEI193 Schwann cells. Int. J. Med. Sci. 2019, 16, 145–155. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Shu, S.; Wang, H.; Zhu, J.; Liu, Z.; Yang, D.; Wu, W.; Cai, J.; Chen, A.; Tang, C.; Dong, Z. Reciprocal regulation between ER stress and autophagy in renal tubular fibrosis and apoptosis. Cell Death Dis. 2021, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H. Renal fibrosis. Korean J. Pediatr. 2010, 53, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Niwa, T. AST-120 ameliorates epithelial-to-mesenchymal transition and interstitial fibrosis in the kidneys of chronic kidney disease rats. J. Ren. Nutr. 2012, 22, 176–180. [Google Scholar] [CrossRef]
- Su, P.Y.; Lee, Y.H.; Kuo, L.N.; Chen, Y.C.; Chen, C.; Kang, Y.N.; Chang, E.H. Efficacy of AST-120 for Patients With Chronic Kidney Disease: A Network Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2021, 12, 676345. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Shimoda, H.; Fukagawa, M. Accumulated uremic toxins attenuate bone mechanical properties in rats with chronic kidney disease. Bone 2013, 57, 477–483. [Google Scholar] [CrossRef]
- Zheng, C.M.; Zheng, J.Q.; Wu, C.C.; Lu, C.L.; Shyu, J.F.; Yung-Ho, H.; Wu, M.Y.; Chiu, I.J.; Wang, Y.H.; Lin, Y.F.; et al. Bone loss in chronic kidney disease: Quantity or quality? Bone 2016, 87, 57–70. [Google Scholar] [CrossRef]
- Kazama, J.J.; Matsuo, K.; Iwasaki, Y.; Fukagawa, M. Chronic kidney disease and bone metabolism. J. Bone Min. Metab. 2015, 33, 245–252. [Google Scholar] [CrossRef]
- Nakada, Y.; Onoue, K.; Nakano, T.; Ishihara, S.; Kumazawa, T.; Nakagawa, H.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; et al. AST-120, an Oral Carbon Absorbent, Protects against the Progression of Atherosclerosis in a Mouse Chronic Renal Failure Model by Preserving sFlt-1 Expression Levels. Sci. Rep. 2019, 9, 15571. [Google Scholar] [CrossRef]
- Ito, S.; Higuchi, Y.; Yagi, Y.; Nishijima, F.; Yamato, H.; Ishii, H.; Osaka, M.; Yoshida, M. Reduction of indoxyl sulfate by AST-120 attenuates monocyte inflammation related to chronic kidney disease. J. Leukoc. Biol. 2013, 93, 837–845. [Google Scholar] [CrossRef]
- Namikoshi, T.; Tomita, N.; Satoh, M.; Sakuta, T.; Kuwabara, A.; Kobayashi, S.; Higuchi, Y.; Nishijima, F.; Kashihara, N. Oral adsorbent AST-120 ameliorates endothelial dysfunction independent of renal function in rats with subtotal nephrectomy. Hypertens. Res. 2009, 32, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol. Dial. Transpl. 2011, 26, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Yoshifuji, A.; Wakino, S.; Irie, J.; Matsui, A.; Hasegawa, K.; Tokuyama, H.; Hayashi, K.; Itoh, H. Oral adsorbent AST-120 ameliorates gut environment and protects against the progression of renal impairment in CKD rats. Clin. Exp. Nephrol. 2018, 22, 1069–1078. [Google Scholar] [CrossRef]
- Hsu, C.K.; Su, S.C.; Chang, L.C.; Yang, K.J.; Lee, C.C.; Hsu, H.J.; Chen, Y.T.; Sun, C.Y.; Wu, I.W. Oral Absorbent AST-120 Is Associated with Compositional and Functional Adaptations of Gut Microbiota and Modification of Serum Short and Medium-Chain Fatty Acids in Advanced CKD Patients. Biomedicines 2022, 10, 2234. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Adenine | Adenine + AST120 | |
|---|---|---|---|
| Serum BUN (mg/d1) | 24.6 (23.3, 27.2) | 172.8 (160.4, 179.7) * | 123.7 (37.6, 146.3) *,# |
| Serum creatinine (mg/dl) | 0.34 (0.27, 0.4) | 1.22 (1.15, 1.44) * | 1.09 (0.4, 1.17) *,# |
| Serum indoxyl (μg/mL) | 1.33 (0.75, 1.88) | 52.8 (42.31, 68.05) * | 36.34 (4.27, 38.6) *,# |
| Renal total iron content (μM) | 0.89 (0.36, 1.11) | 1.41 (0.92, 2.17) * | 0.78 (0.71, 1.27) # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, L.-T.; Weng, T.-I.; Chang, T.-Y.; Lan, K.-C.; Chiang, C.-K.; Liu, S.-H. Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo. Antioxidants 2023, 12, 1931. https://doi.org/10.3390/antiox12111931
Tsai L-T, Weng T-I, Chang T-Y, Lan K-C, Chiang C-K, Liu S-H. Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo. Antioxidants. 2023; 12(11):1931. https://doi.org/10.3390/antiox12111931
Chicago/Turabian StyleTsai, Li-Ting, Te-I Weng, Ting-Yu Chang, Kuo-Cheng Lan, Chih-Kang Chiang, and Shing-Hwa Liu. 2023. "Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo" Antioxidants 12, no. 11: 1931. https://doi.org/10.3390/antiox12111931
APA StyleTsai, L. -T., Weng, T. -I., Chang, T. -Y., Lan, K. -C., Chiang, C. -K., & Liu, S. -H. (2023). Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo. Antioxidants, 12(11), 1931. https://doi.org/10.3390/antiox12111931