
GYY4137-Derived Hydrogen Sulfide Donates Electrons to the Mitochondrial Electron Transport Chain via Sulfide: Quinone Oxidoreductase in Endothelial Cells

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Sulfide Solution, Preparation, and Use
2.3. Oxygen Consumption Rate Seahorse
2.4. Analysing the Presence of SQOR by Western Blot
2.5. Measurement of Cellular Respiration and Sulfide Oxidation
2.6. Imaging
2.7. AzMC Dose Response
2.8. Lead Acetate
2.9. Electron Microscopy
2.10. Data Analysis
3. Results
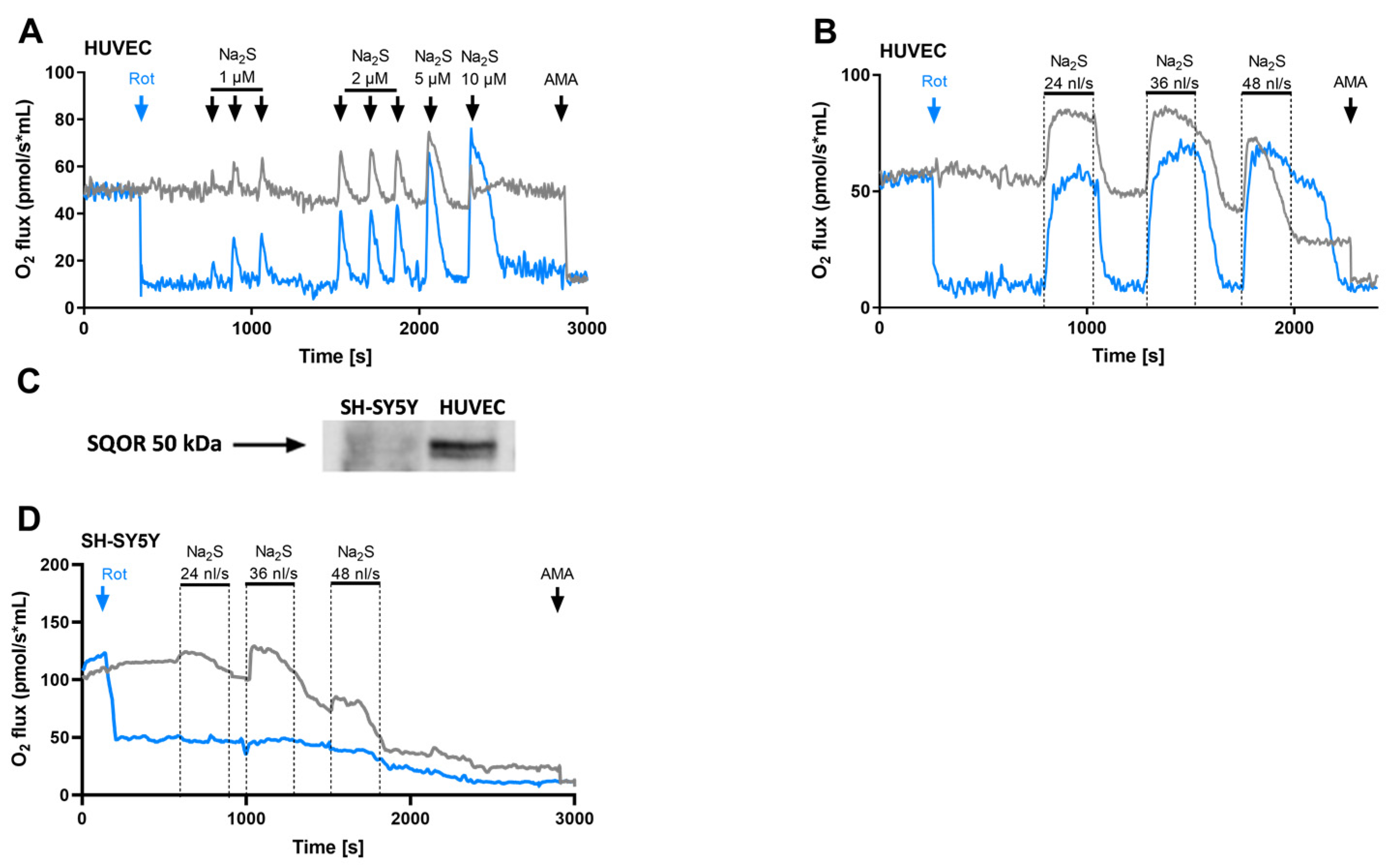
3.1. Human Umbilical Vein Endothelial Cells Oxidize H2S via the Sulfide:Quinone Oxidoreductase
3.2. Stoichiometry between H2S Oxidation and Oxygen Consumption
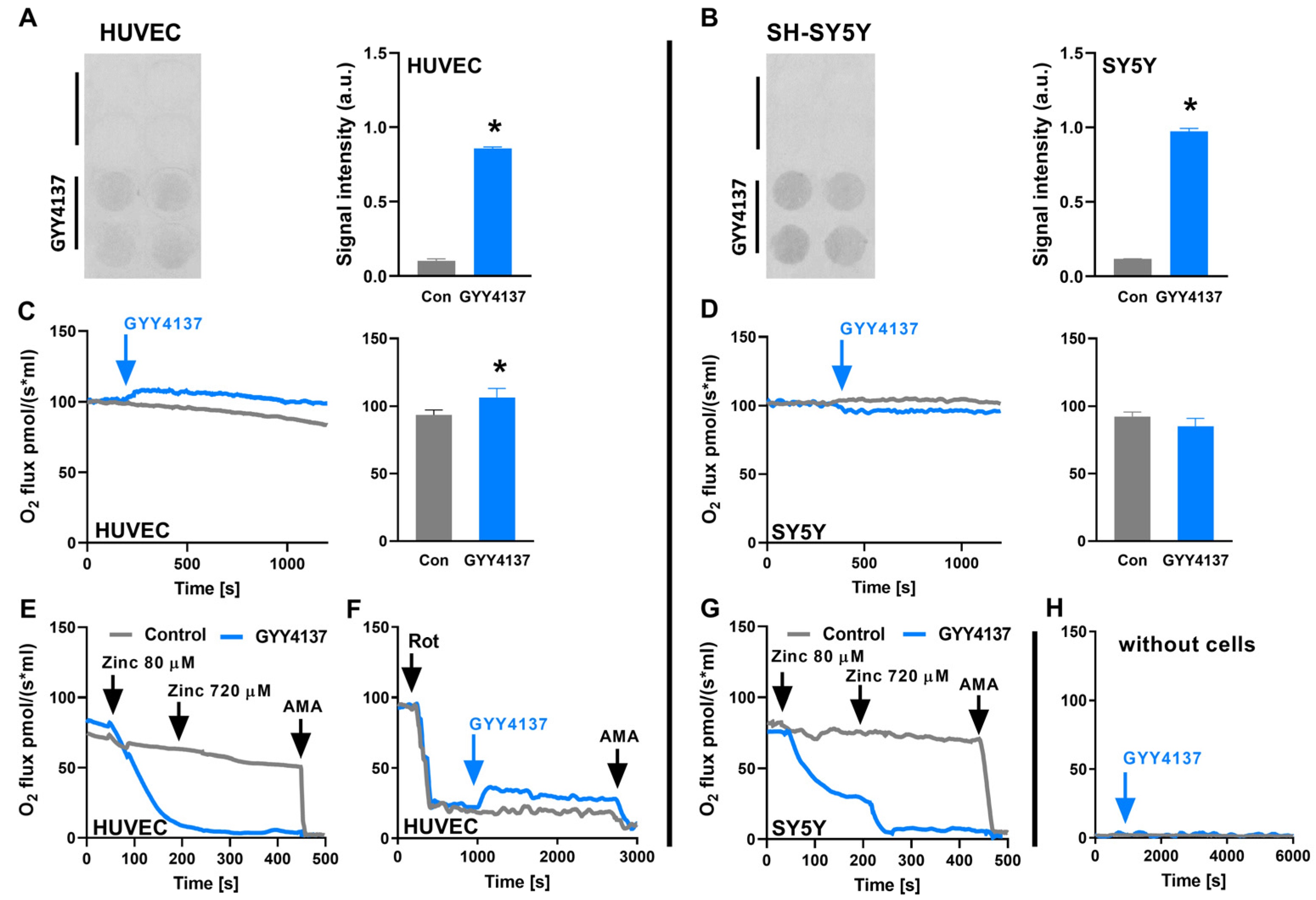
3.3. SQOR Oxidises H2S Released from GYY4137
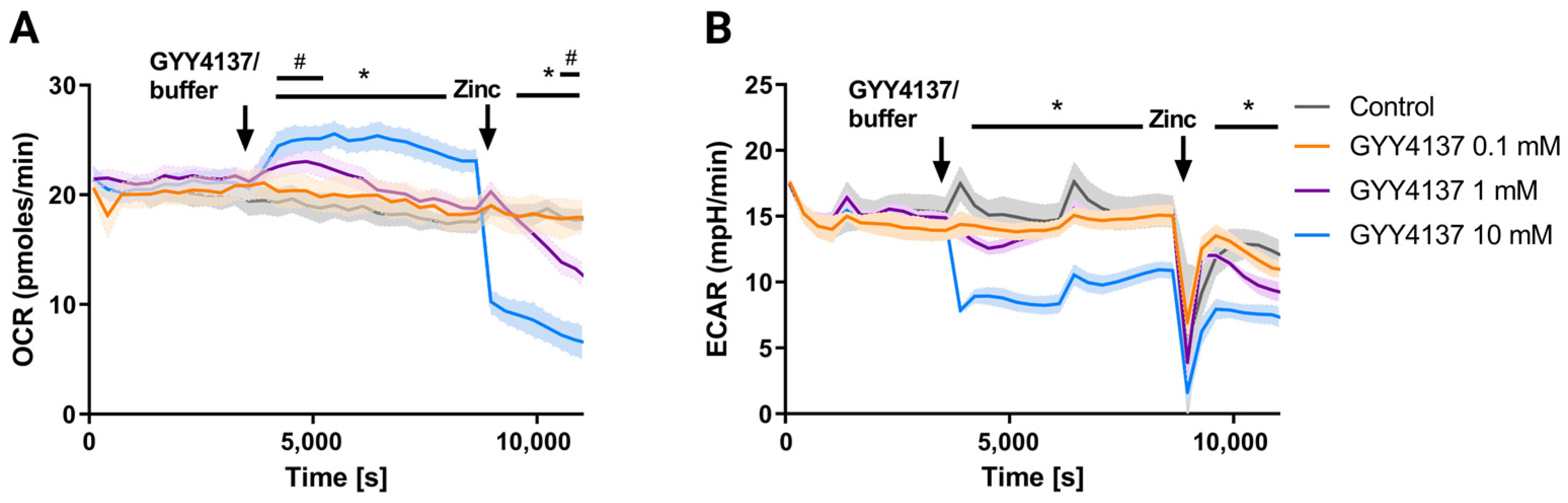
3.4. GYY4137 Only Increases Oxygen Consumption in Cells with a SQOR
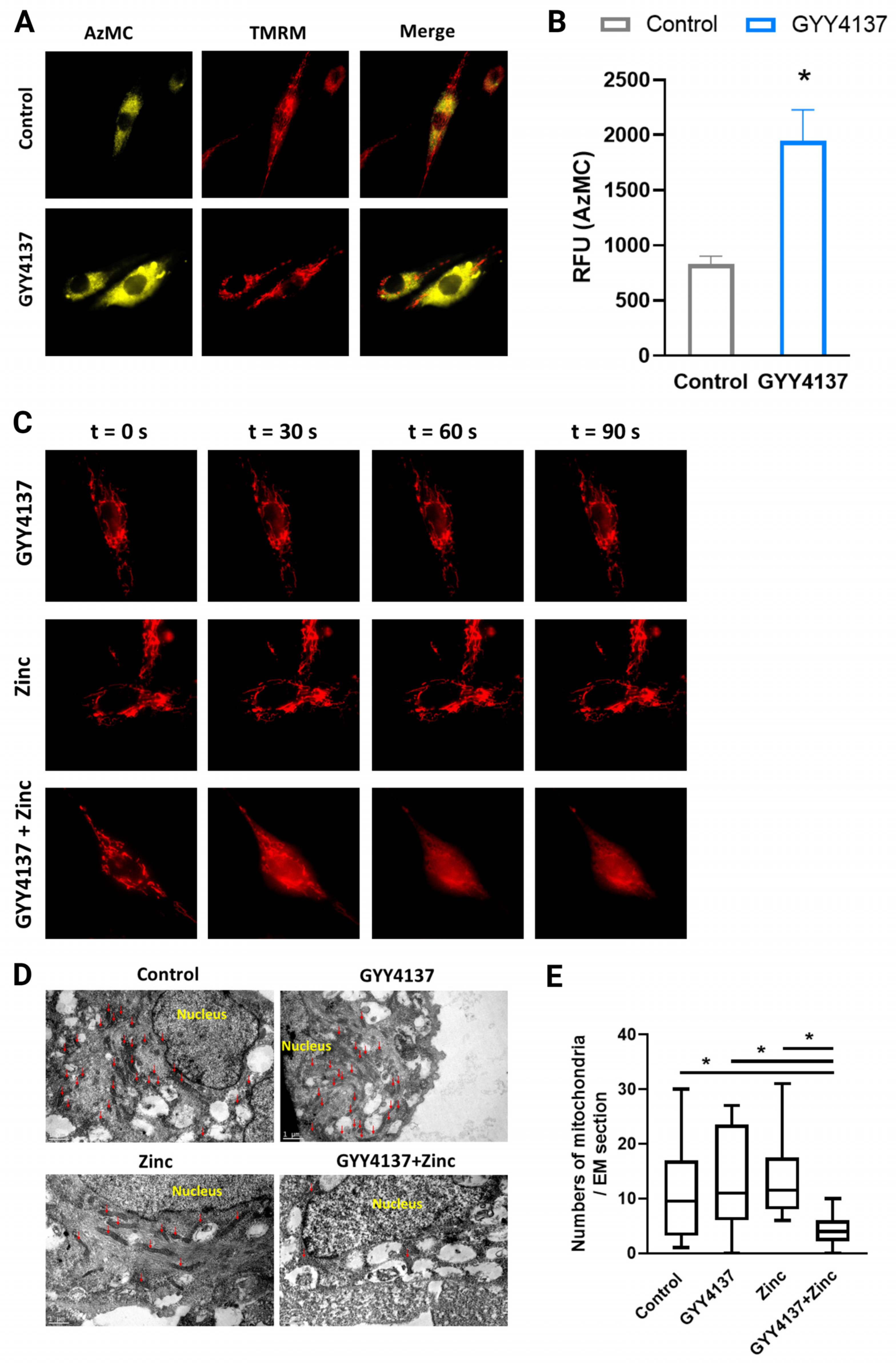
3.5. GYY4137 Increased Intramitochondrial H2S Levels
4. Discussion
4.1. Sulfide Oxidation by Human Endothelial Cells
4.2. H2S Donation by GYY4137 Influences Mitochondrial Oxygen Consumption
4.3. GYY4137 Administration and Consequences of Intracellular (Autocrine) H2S Release
4.4. Effects of H2S on Protein Function
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johansen, D.; Ytrehus, K.; Baxter, G.F. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury—Evidence for a role of K ATP channels. Basic Res. Cardiol. 2006, 101, 53–60. [Google Scholar] [CrossRef]
- Guo, W.; Cheng, Z.Y.; Zhu, Y.Z. Hydrogen sulfide and translational medicine. Acta Pharmacol. Sin. 2013, 34, 1284–1291. [Google Scholar] [CrossRef]
- Tao, L.; Yu, Q.; Zhao, P.; Yang, Q.; Wang, B.; Yang, Y.; Kuai, J.; Ding, Q. Preconditioning with hydrogen sulfide ameliorates cerebral ischemia/reperfusion injury in a mouse model of transient middle cerebral artery occlusion. Chem. Biol. Interact 2019, 310, 108738. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef]
- Abou-Hamdan, A.; Guedouari-Bounihi, H.; Lenoir, V.; Andriamihaja, M.; Blachier, F.; Bouillaud, F. Oxidation of H2S in mammalian cells and mitochondria. Methods Enzym. 2015, 554, 201–228. [Google Scholar]
- Sonobe, T.; Haouzi, P. Sulfide Intoxication-Induced Circulatory Failure is Mediated by a Depression in Cardiac Contractility. Cardiovasc. Toxicol. 2016, 16, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Tvedt, B.; Skyberg, K.; Aaserud, O.; Hobbesland, A.; Mathiesen, T. Brain damage caused by hydrogen sulfide: A follow-up study of six patients. Am. J. Ind. Med. 1991, 20, 91–101. [Google Scholar] [CrossRef]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Salto-Tellez, M.; Tan, C.H.; Whiteman, M.; Moore, P.K. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic. Biol. Med. 2009, 47, 103–113. [Google Scholar] [CrossRef]
- Chen, L.J.; Ning, J.Z.; Cheng, F.; Rao, T.; Yu, W.M.; Ruan, Y.; Wu, J.F.; Li, R.G.; Geng, R.X. Comparison of Intraperitoneal and Intratesticular GYY4137 Therapy for the Treatment of Testicular Ischemia Reperfusion Injury in Rats. Curr. Med. Sci. 2020, 40, 332–338. [Google Scholar] [CrossRef]
- Cui, N.; Luo, H.; Zhao, Y. Protective effect of GYY4137, a water-soluble hydrogen sulfide-releasing molecule, on intestinal ischemia-reperfusion. Mol. Med. Rep. 2020, 21, 1633–1639. [Google Scholar] [CrossRef]
- Chen, S.; Bu, D.; Ma, Y.; Zhu, J.; Sun, L.; Zuo, S.; Ma, J.; Li, T.; Chen, Z.; Zheng, Y.; et al. GYY4137 ameliorates intestinal barrier injury in a mouse model of endotoxemia. Biochem. Pharmacol. 2016, 118, 59–67. [Google Scholar] [CrossRef]
- Zheng, Q.; Pan, L.; Ji, Y. H2S protects against diabetes-accelerated atherosclerosis by preventing the activation of NLRP3 inflammasome. J. Biomed. Res. 2019, 34, 94–102. [Google Scholar] [CrossRef]
- Zheng, Y.; Lv, P.; Huang, J.; Ke, J.; Yan, J. GYY4137 exhibits anti-atherosclerosis effect in apolipoprotein E (−/−) mice via PI3K/Akt and TLR4 signalling. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1231–1239. [Google Scholar] [CrossRef]
- Lobb, I.; Jiang, J.; Lian, D.; Liu, W.; Haig, A.; Saha, M.N.; Torregrossa, R.; Wood, M.E.; Whiteman, M.; Sener, A. Hydrogen Sulfide Protects Renal Grafts Against Prolonged Cold Ischemia-Reperfusion Injury via Specific Mitochondrial Actions. Am. J. Transplant. 2017, 17, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. 2019, 33, 13098–13125. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P.; Martinez, J.; Miller, B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 2011, 13, e19. [Google Scholar] [CrossRef]
- Star, B.S.; Boahen, C.K.; van der Slikke, E.C.; Quinten, V.M.; Ter Maaten, J.C.; Henning, R.H.; Kumar, V.; Bouma, H.R. Plasma proteomic characterization of the development of acute kidney injury in early sepsis patients. Sci. Rep. 2022, 12, 19705. [Google Scholar] [CrossRef]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascon, G.A.; Hernandez, G.; Murray, P.; De Backer, D.; Workgroup, A.X. The Endothelium in Sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Kubitza, M.; Hauska, G.; Pina, A.L. The vertebrate homologue of sulfide-quinone reductase in mammalian mitochondria. Cell Tissue Res. 2014, 358, 779–792. [Google Scholar] [CrossRef]
- Ackermann, M.; Kubitza, M.; Maier, K.; Brawanski, A.; Hauska, G.; Pina, A.L. The vertebrate homolog of sulfide-quinone reductase is expressed in mitochondria of neuronal tissues. Neuroscience 2011, 199, 1–12. [Google Scholar] [CrossRef]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, S.; Minamino, S.; Takase, M.; Ishiyama, Y.; Hosokura, H.; Kohda, T.; Ikeda, Y.; Fujimoto, Y. Hydrogen sulfide donor GYY4137 suppresses proliferation of human colorectal cancer Caco-2 cells by inducing both cell cycle arrest and cell death. Heliyon 2019, 5, e02244. [Google Scholar] [CrossRef] [Green Version]
- Sestito, S.; Daniele, S.; Pietrobono, D.; Citi, V.; Bellusci, L.; Chiellini, G.; Calderone, V.; Martini, C.; Rapposelli, S. Memantine prodrug as a new agent for Alzheimer’s Disease. Sci. Rep. 2019, 9, 4612. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [Green Version]
- Abramavicius, S.; Petersen, A.G.; Renaltan, N.S.; Prat-Duran, J.; Torregrossa, R.; Stankevicius, E.; Whiteman, M.; Simonsen, U. GYY4137 and Sodium Hydrogen Sulfide Relaxations Are Inhibited by L-Cysteine and K(V)7 Channel Blockers in Rat Small Mesenteric Arteries. Front. Pharmacol. 2021, 12, 613989. [Google Scholar] [CrossRef]
- Pal, V.K.; Agrawal, R.; Rakshit, S.; Shekar, P.; Murthy, D.T.N.; Vyakarnam, A.; Singh, A. Hydrogen sulfide blocks HIV rebound by maintaining mitochondrial bioenergetics and redox homeostasis. Elife 2021, 10, e68487. [Google Scholar] [CrossRef]
- Bouillaud, F. Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio. Biomolecules 2022, 12, 361. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Modis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [Green Version]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide catabolism ameliorates hypoxic brain injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2020, 31, 207. [Google Scholar] [CrossRef] [Green Version]
- Roorda, M.; Miljkovic, J.L.; van Goor, H.; Henning, R.H.; Bouma, H.R. Spatiotemporal regulation of hydrogen sulfide signaling in the kidney. Redox Biol. 2021, 43, 101961. [Google Scholar] [CrossRef]
- Stein, A.; Bailey, S.M. Redox Biology of Hydrogen Sulfide: Implications for Physiology, Pathophysiology, and Pharmacology. Redox Biol. 2013, 1, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Modis, K.; Panopoulos, P.; Coletta, C.; Papapetropoulos, A.; Szabo, C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem. Pharmacol. 2013, 86, 1311–1319. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline | 24 nL/s Na2S | 36 nL/s Na2S | 48 nL/s Na2S | Stoichiometry @24 nL/s | |
|---|---|---|---|---|---|
| O2 pmol/s × mL | O2 pmol/s × mL | O2 pmol/s × mL | O2 pmol/s × mL | ∆JO2/JNa2S | |
| Control | 55 | 83 | 83 | 72 | 0.47 |
| Rotenone | 10 | 50 | 68 | 68 | 0.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Star, B.S.; van der Slikke, E.C.; Ransy, C.; Schmitt, A.; Henning, R.H.; Bouillaud, F.; Bouma, H.R. GYY4137-Derived Hydrogen Sulfide Donates Electrons to the Mitochondrial Electron Transport Chain via Sulfide: Quinone Oxidoreductase in Endothelial Cells. Antioxidants 2023, 12, 587. https://doi.org/10.3390/antiox12030587
Star BS, van der Slikke EC, Ransy C, Schmitt A, Henning RH, Bouillaud F, Bouma HR. GYY4137-Derived Hydrogen Sulfide Donates Electrons to the Mitochondrial Electron Transport Chain via Sulfide: Quinone Oxidoreductase in Endothelial Cells. Antioxidants. 2023; 12(3):587. https://doi.org/10.3390/antiox12030587
Chicago/Turabian StyleStar, Bastiaan S., Elisabeth C. van der Slikke, Céline Ransy, Alain Schmitt, Robert H. Henning, Frédéric Bouillaud, and Hjalmar R. Bouma. 2023. "GYY4137-Derived Hydrogen Sulfide Donates Electrons to the Mitochondrial Electron Transport Chain via Sulfide: Quinone Oxidoreductase in Endothelial Cells" Antioxidants 12, no. 3: 587. https://doi.org/10.3390/antiox12030587
APA StyleStar, B. S., van der Slikke, E. C., Ransy, C., Schmitt, A., Henning, R. H., Bouillaud, F., & Bouma, H. R. (2023). GYY4137-Derived Hydrogen Sulfide Donates Electrons to the Mitochondrial Electron Transport Chain via Sulfide: Quinone Oxidoreductase in Endothelial Cells. Antioxidants, 12(3), 587. https://doi.org/10.3390/antiox12030587