

Neuroimaging in Primary Coenzyme-Q10-Deficiency Disorders

 , , , , ,
, , , , , 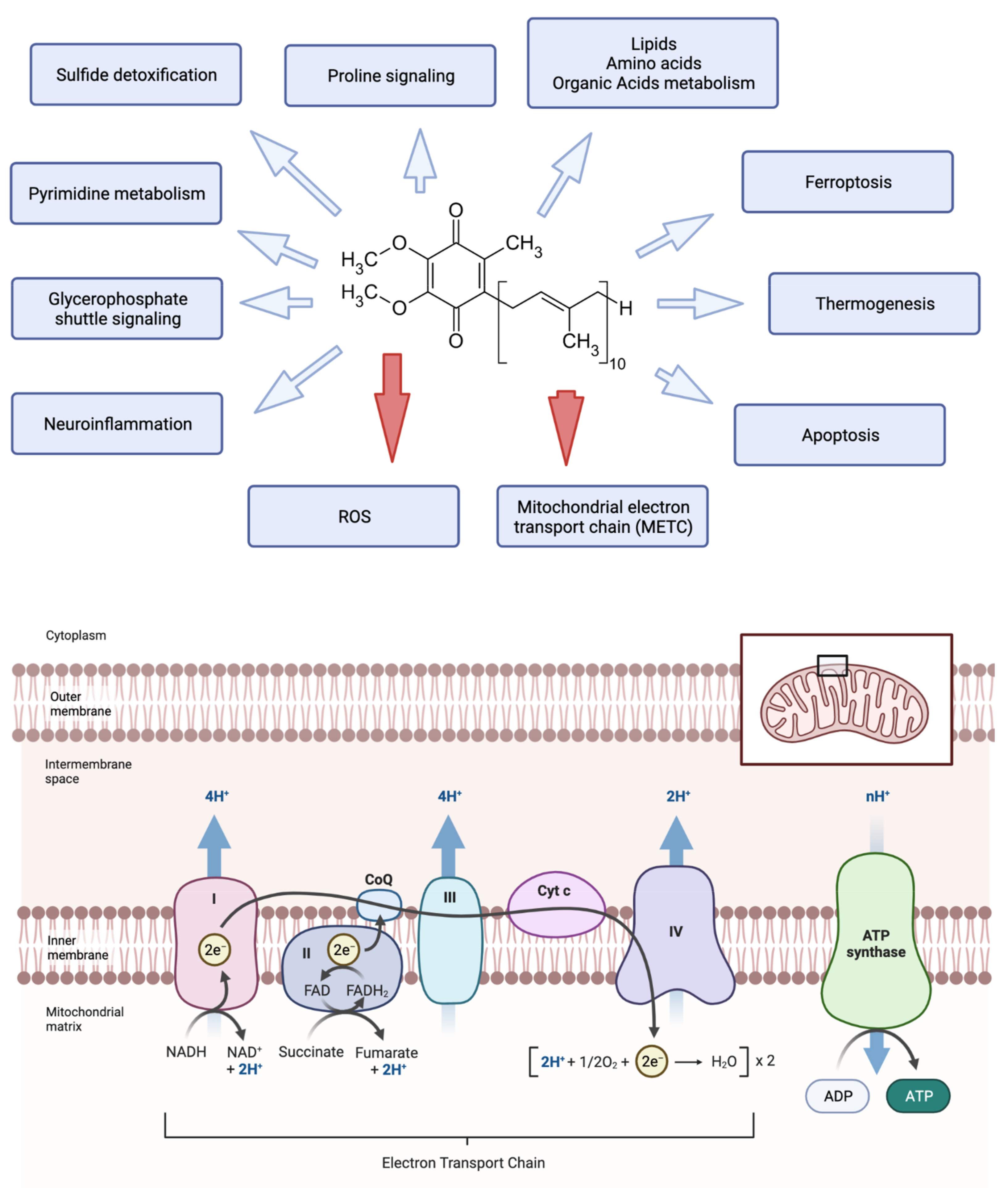{kind=link}
{kind=link}
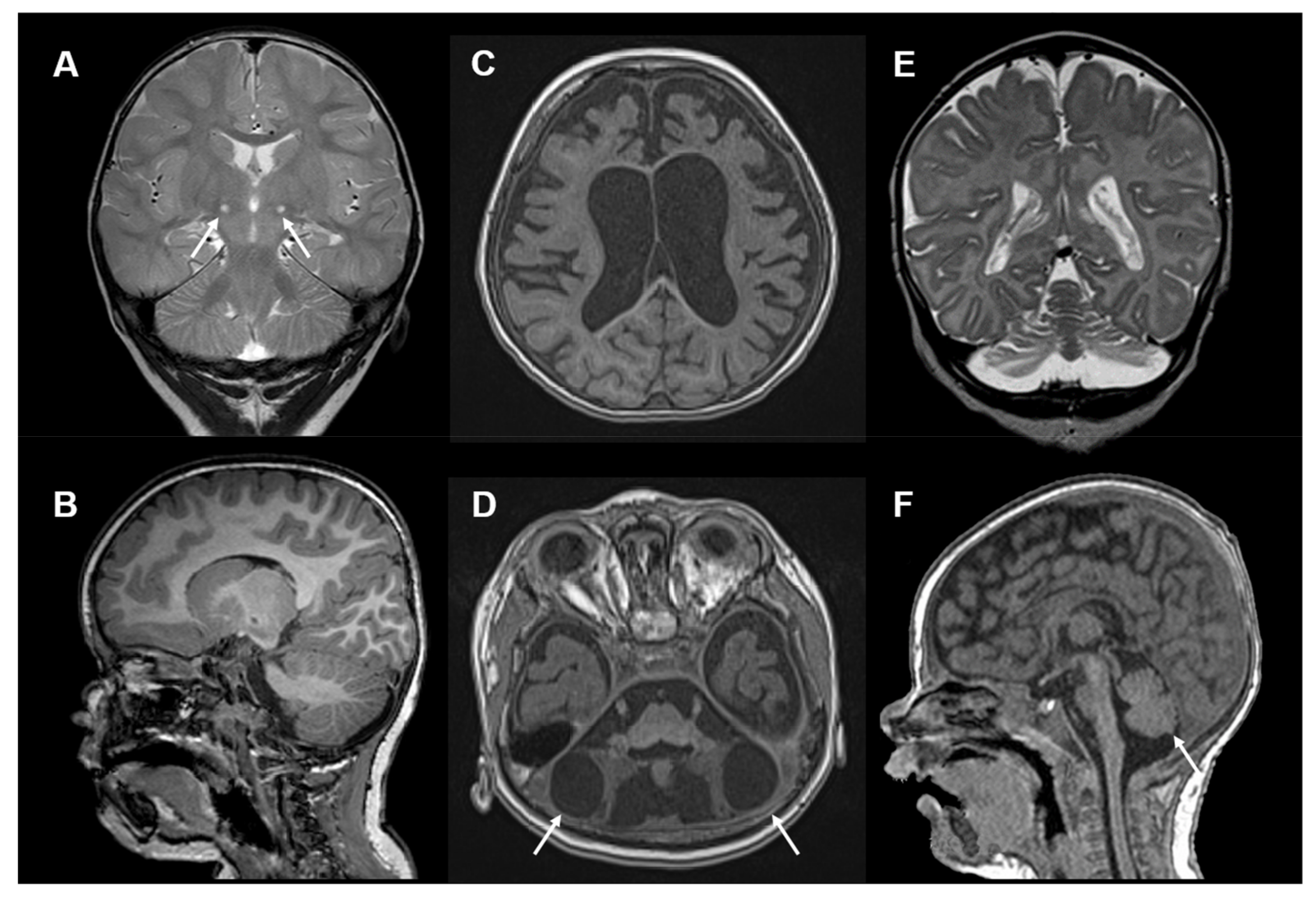{kind=link}
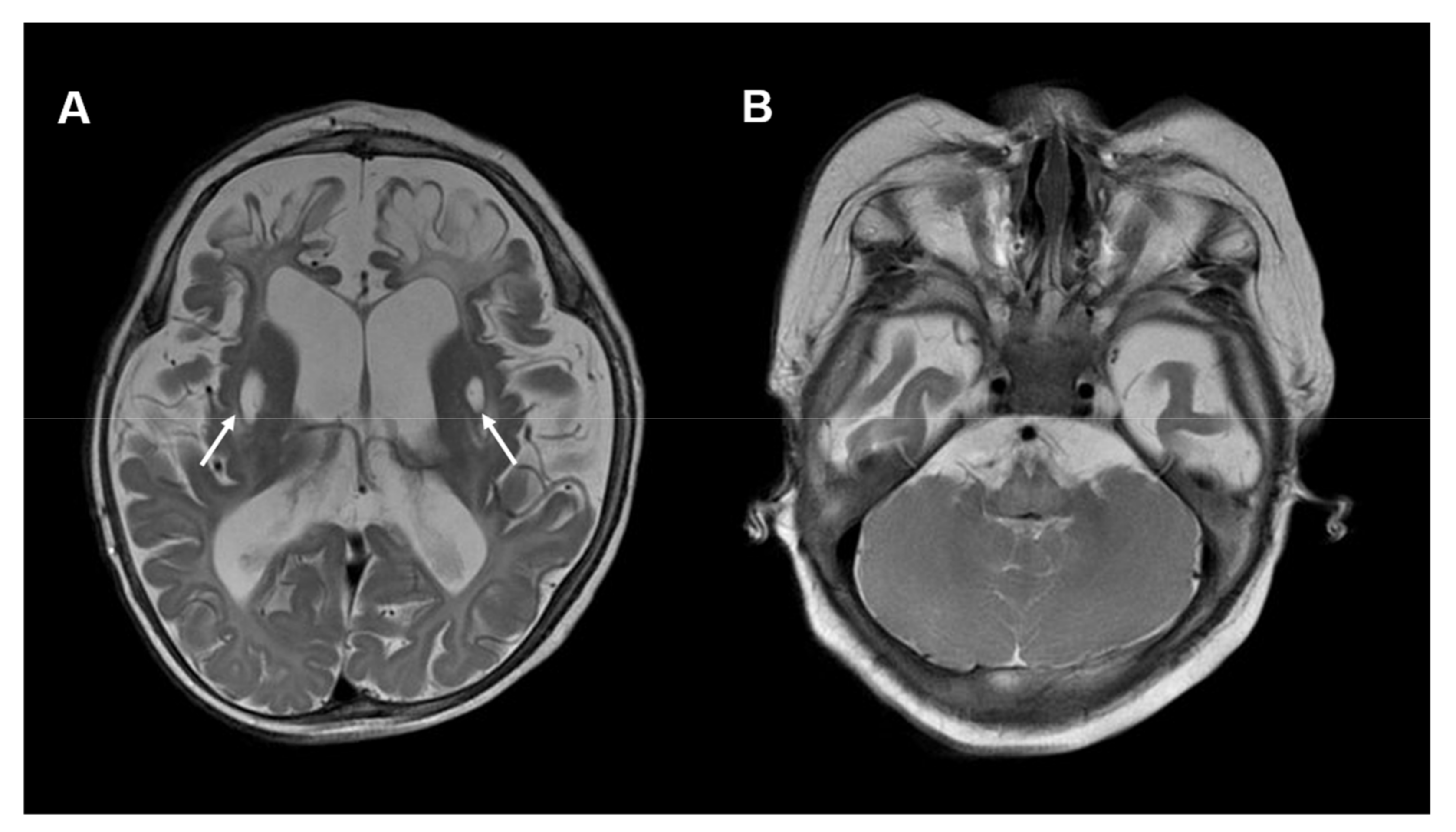{kind=link}
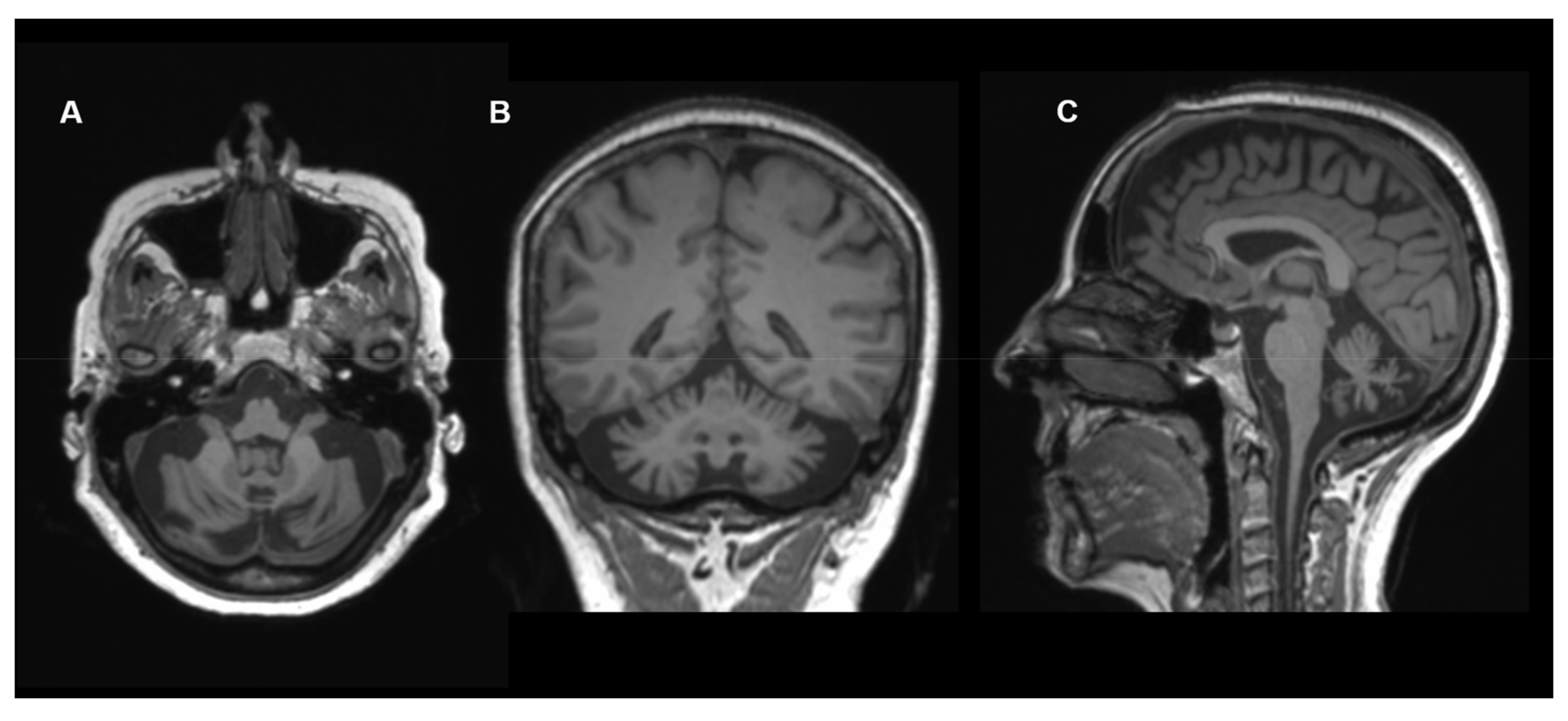{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. PDSS1 Deficiency
3.2. PDSS2 Deficiency
3.3. COQ2 Deficiency
3.4. COQ4 Deficiency
3.5. COQ5 Deficiency
3.6. COQ6 Deficiency
3.7. COQ7 Deficiency
3.8. COQ8A Deficiency
3.9. COQ8B Deficiency
3.10. COQ9 Deficiency
3.11. HPDL Deficiency
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhagavan, H.N.; Chopra, R. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40 Pt 3, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q–biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q(10) Deficiency. In GeneReviews(®); Adam, M., Everman, D., Mirzaa, G., Pagon, R., Wallace, S., Bean, L.J., Gripp, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Banh, R.S.; Kim, E.S.; Spillier, Q.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Shi, G.; Jones, D.R.; Kimmelman, A.C.; Pacold, M.E. The polar oxy-metabolome reveals the 4-hydroxymandelate CoQ10 synthesis pathway. Nature 2021, 597, 420–425. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Hidalgo-Gutierrez, A.; Kleiner, G.; Lopez, L.C. The Role of Sulfide Oxidation Impairment in the Pathogenesis of Primary CoQ Deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef]
- González-García, P.; Díaz-Casado, M.E.; Hidalgo-Gutiérrez, A.; Jiménez-Sánchez, L.; Bakkali, M.; Barriocanal-Casado, E.; Escames, G.; Chiozzi, R.Z.; Völlmy, F.; Zaal, E.A.; et al. The Q-junction and the inflammatory response are critical pathological and therapeutic factors in CoQ deficiency. Redox Biol. 2022, 55, 102403. [Google Scholar] [CrossRef]
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef]
- Gu, R.; Zhang, F.; Chen, G.; Han, C.; Liu, J.; Ren, Z.; Zhu, Y.; Waddington, J.L.; Zheng, L.T.; Zhen, X. Clk1 deficiency promotes neuroinflammation and subsequent dopaminergic cell death through regulation of microglial metabolic reprogramming. Brain Behav. Immun. 2017, 60, 206–219. [Google Scholar] [CrossRef]
- Duberley, K.E.C.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q 10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2013, 36, 63–73. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef]
- Nardecchia, F.; De Giorgi, A.; Palombo, F.; Fiorini, C.; De Negri, A.M.; Carelli, V.; Caporali, L.; Leuzzi, V. Missense PDSS1 mutations in CoenzymeQ10 synthesis cause optic atrophy and sensorineural deafness. Ann. Clin. Transl. Neurol. 2021, 8, 247–251. [Google Scholar] [CrossRef]
- Vasta, V.; Lawrence Merritt, J., II; Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum. Pediatr. Int. 2012, 54, 585–601. [Google Scholar] [CrossRef]
- Bellusci, M.; García-Silva, M.T.; de Aragón, A.M.; Martín, M.A. Distal phalangeal erythema in an infant with biallelic PDSS1 mutations: Expanding the phenotype of primary Coenzyme Q 10 deficiency. JIMD Rep. 2021, 62, 3–5. [Google Scholar] [CrossRef]
- Rötig, A.; Appelkvist, E.-L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Iványi, B.; Rácz, G.Z.; Gál, P.; Brinyiczki, K.; Bódi, I.; Kalmár, T.; Maróti, Z.; Bereczki, C. Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr. Nephrol. 2018, 33, 439–446. [Google Scholar] [CrossRef]
- Schijvens, A.M.; van de Kar, N.C.; Bootsma-Robroeks, C.M.; Cornelissen, E.A.; Heuvel, L.P.V.D.; Schreuder, M.F. Mitochondrial Disease and the Kidney With a Special Focus on CoQ10 Deficiency. Kidney Int. Rep. 2020, 5, 2146–2159. [Google Scholar] [CrossRef]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Hirano, M.; DiMauro, S. Mutant COQ2 in multiple-system atrophy. N. Engl. J. Med. 2014, 371, 81–82. [Google Scholar] [PubMed]
- Hashemi, S.S.; Zare-Abdollahi, D.; Bakhshandeh, M.K.; Vafaee, A.; Abolhasani, S.; Inanloo Rahatloo, K.; DanaeeFard, F.; Farboodi, N.; Rohani, M.; Alavi, A. Clinical spectrum in multiple families with primary COQ(10) deficiency. Am. J. Med. Genet. A 2021, 185, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C.; et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatr. Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- Casarin, A.; Jimenez-Ortega, J.C.; Trevisson, E.; Pertegato, V.; Doimo, M.; Ferrero-Gomez, M.L.; Abbadi, S.; Artuch, R.; Quinzii, C.; Hirano, M.; et al. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochem. Biophys. Res. Commun. 2008, 372, 35–39. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta 2009, 1791, 69–75. [Google Scholar] [CrossRef]
- Calvo, G.T.B.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S.; et al. COQ4 Mutations Cause a Broad Spectrum of Mitochondrial Disorders Associated with CoQ10 Deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef]
- Laugwitz, L.; Seibt, A.; Herebian, D.; Peralta, S.; Kienzle, I.; Buchert, R.; Falb, R.; Gauck, D.; Müller, A.; Grimmel, M.; et al. Human COQ4 deficiency: Delineating the clinical, metabolic and neuroimaging phenotypes. J. Med. Genet. 2022, 59, 878–887. [Google Scholar] [CrossRef]
- Cordts, I.; Semmler, L.; Prasuhn, J.; Seibt, A.; Herebian, D.; Navaratnarajah, T.; Park, J.; Deininger, N.; Laugwitz, L.; Göricke, S.L.; et al. Bi-Allelic COQ4 Variants Cause Adult-Onset Ataxia-Spasticity Spectrum Disease. Mov. Disord. 2022, 37, 2147–2153. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta 2014, 1841, 1628–1638. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Justine Perrin, R.; Rousset-Rouvière, C.; Garaix, F.; Cano, A.; Conrath, J.; Boyer, O.; Tsimaratos, M. COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy. JIMD Rep. 2020, 54, 37–44. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Wang, N.; Zheng, Y.; Zhang, L.; Tian, X.; Fang, Y.; Qi, M.; Du, J.; Chen, S.; Chen, S.; Li, J.; et al. A Family Segregating Lethal Primary Coenzyme Q10 Deficiency Due to Two Novel COQ6 Variants. Front. Genet. 2021, 12, 811833. [Google Scholar] [CrossRef]
- Jacquier, A.; Theuriet, J.; Fontaine, F.; Mosbach, V.; Lacoste, N.; Ribault, S.; Risson, V.; Carras, J.; Coudert, L.; Simonet, T.; et al. Homozygous COQ7 mutation: A new cause of potentially treatable distal hereditary motor neuropathy. Brain, 2022; ahead of print. [Google Scholar] [CrossRef]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Wang, Y.; Gumus, E.; Hekimi, S. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol. Genet. Metab. Rep. 2022, 31, 100877. [Google Scholar] [CrossRef]
- Kwong, A.K.; Chiu, A.T.; Tsang, M.H.; Lun, K.S.; Rodenburg, R.J.T.; Smeitink, J.; Chung, B.H.; Fung, C.W. A fatal case of COQ7-associated primary coenzyme Q(10) deficiency. JIMD Rep. 2019, 47, 23–29. [Google Scholar] [CrossRef]
- Xie, L.X.; Hsieh, E.J.; Watanabe, S.; Allan, C.M.; Chen, J.Y.; Tran, U.C.; Clarke, C.F. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim. Biophys. Acta 2011, 1811, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ashizawa, T.; Peng, D. Primary coenzyme Q10 deficiency due to COQ8A gene mutations. Mol. Genet. Genom. Med. 2020, 8, e1420. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Traschütz, A.; Schirinzi, T.; Laugwitz, L.; Murray, N.H.; Bingman, C.A.; Reich, S.; Kern, J.; Heinzmann, A.; Vasco, G.; Bertini, E.; et al. Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients. Ann. Neurol. 2020, 88, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Galosi, S.; Barca, E.; Carrozzo, R.; Schirinzi, T.; Quinzii, C.M.; Lieto, M.; Vasco, G.; Zanni, G.; Di Nottia, M.; Galatolo, D.; et al. Dystonia-Ataxia with early handwriting deterioration in COQ8A mutation carriers: A case series and literature review. Park. Relat. Disord. 2019, 68, 8–16. [Google Scholar] [CrossRef]
- Ashrafi, M.R.; Haghighi, R.; Badv, R.S.; Ghabeli, H.; Tavasoli, A.R.; Pourbakhtyaran, E.; Rezaei, Z.; Mahdieh, N.; Mohammadi, P.; Heidari, M. Epilepsia Partialis Continua a Clinical Feature of a Missense Variant in the ADCK3 Gene and Poor Response to Therapy. J. Mol. Neurosci. 2022, 72, 1125–1132. [Google Scholar] [CrossRef]
- Değerliyurt, A.; Gülleroğlu, N.B.; Kibar Gül, A.E. Primary CoQ(10) deficiency with a severe phenotype due to the c.901 C > T (p.R301W) mutation in the COQ8A gene. Int. J. Neurosci. 2022; ahead of print. [Google Scholar] [CrossRef]
- Paprocka, J.; Nowak, M.; Chuchra, P.; Śmigiel, R. COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency. Metabolites 2022, 12, 955. [Google Scholar] [CrossRef]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta 2016, 1857, 1079–1085. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Zhai, S.-B.; Zhang, L.; Sun, B.-C.; Zhang, Y.; Ma, Q.-S. Early-onset COQ8B (ADCK4) glomerulopathy in a child with isolated proteinuria: A case report and literature review. BMC Nephrol. 2020, 21, 406. [Google Scholar] [CrossRef]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sánchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001, 139, 456–458. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef]
- Danhauser, K.; Herebian, D.; Haack, T.B.; Rodenburg, R.J.; Strom, T.M.; Meitinger, T.; Klee, D.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur. J. Hum. Genet. 2016, 24, 450–454. [Google Scholar] [CrossRef]
- Smith, A.C.; Ito, Y.; Ahmed, A.; Schwartzentruber, J.A.; Beaulieu, C.L.; Aberg, E.; Majewski, J.; Bulman, D.E.; Horsting-Wethly, K.; Koning, D.V.-D.; et al. A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9. J. Inherit. Metab. Dis. 2018, 41, 719–729. [Google Scholar] [CrossRef]
- Olgac, A.; Öztoprak, Ü.; Kasapkara, S.; Kılıç, M.; Yüksel, D.; Derinkuyu, E.B.; Yıldız, Y.T.; Ceylaner, S.; Ezgu, F.S. A rare case of primary coenzyme Q10 deficiency due to COQ9 mutation. J. Pediatr. Endocrinol. Metab. 2020, 33, 165–170. [Google Scholar] [CrossRef]
- Ghosh, S.G.; Lee, S.; Fabunan, R.; Chai, G.; Zaki, M.S.; Abdel-Salam, G.; Sultan, T.; Ben-Omran, T.; Alvi, J.R.; McEvoy-Venneri, J.; et al. Biallelic variants in HPDL, encoding 4-hydroxyphenylpyruvate dioxygenase-like protein, lead to an infantile neurodegenerative condition. Genet. Med. 2021, 23, 524–533. [Google Scholar] [CrossRef]
- Husain, R.A.; Grimmel, M.; Wagner, M.; Hennings, J.C.; Marx, C.; Feichtinger, R.G.; Saadi, A.; Rostásy, K.; Radelfahr, F.; Bevot, A.; et al. Bi-allelic HPDL Variants Cause a Neurodegenerative Disease Ranging from Neonatal Encephalopathy to Adolescent-Onset Spastic Paraplegia. Am. J. Hum. Genet. 2020, 107, 364–373. [Google Scholar] [CrossRef]
- Wiessner, M.; Maroofian, R.; Ni, M.-Y.; Pedroni, A.; Müller, J.S.; Stucka, R.; Beetz, C.; Efthymiou, S.; Santorelli, F.M.; A Alfares, A.; et al. Biallelic variants in HPDL cause pure and complicated hereditary spastic paraplegia. Brain 2021, 144, 1422–1434. [Google Scholar] [CrossRef]
- Numata-Uematsu, Y.; Uematsu, M.; Yamamoto, T.; Saitsu, H.; Katata, Y.; Oikawa, Y.; Saijyo, N.; Inui, T.; Murayama, K.; Ohtake, A.; et al. Leigh syndrome-like MRI changes in a patient with biallelic HPDL variants treated with ketogenic diet. Mol. Genet. Metab. Rep. 2021, 29, 100800. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, X.; Feng, C.; Fan, X.; Liu, L.; Guo, P.; Lei, Z.; Mei, S. HPDL mutations identified by exome sequencing are associated with infant neurodevelopmental disorders. Mol. Genet. Genom. Med. 2022, 10, e2025. [Google Scholar] [CrossRef] [PubMed]
- Micule, I.; Lace, B.; Wright, N.T.; Chrestian, N.; Strautmanis, J.; Diriks, M.; Stavusis, J.; Kidere, D.; Kleina, E.; Zdanovica, A.; et al. Case Report: Two Families With HPDL Related Neurodegeneration. Front. Genet. 2022, 13, 780764. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Rodrigues, M.; Loureiro, T. Prenatal Diagnosis of Lissencephaly Associated with Biallelic Pathologic Variants in the COQ2 Gene. Acta Med. Port. 2022; ahead of print. [Google Scholar]
- Jurkute, N.; Cancellieri, F.; Pohl, L.; Li, C.H.Z.; Heaton, R.A.; Reurink, J.; Bellingham, J.; Quinodoz, M.; Yioti, G.; Stefaniotou, M.; et al. Biallelic variants in coenzyme Q10 biosynthesis pathway genes cause a retinitis pigmentosa phenotype. NPJ Genom. Med. 2022, 7, 60. [Google Scholar] [CrossRef]
- Lu, S.; Lu, L.-Y.; Liu, M.-F.; Yuan, Q.-J.; Sham, M.-H.; Guan, X.-Y.; Huang, J.-D. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol. Dis. 2012, 45, 219–233. [Google Scholar] [CrossRef]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef]
- Bosch, A.M.; Kamsteeg, E.-J.; Rodenburg, R.J.; van Deutekom, A.W.; Buis, D.R.; Engelen, M.; Cobben, J.-M. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef]
- Będkowska, N.; Zontek, A.; Paprocka, J. Stroke-like Episodes in Inherited Neurometabolic Disorders. Metabolites 2022, 12, 929. [Google Scholar] [CrossRef]
- Hyung, S.; Lee, B.Y.; Park, J.-C.; Kim, J.; Hur, E.-M.; Suh, J.-K.F. Coculture of Primary Motor Neurons and Schwann Cells as a Model for In Vitro Myelination. Sci. Rep. 2015, 5, 15122. [Google Scholar] [CrossRef]
- Ota, K.; Obayashi, M.; Ozaki, K.; Ichinose, S.; Kakita, A.; Tada, M.; Takahashi, H.; Ando, N.; Eishi, Y.; Mizusawa, H.; et al. Relocation of p25α/tubulin polymerization promoting protein from the nucleus to the perinuclear cytoplasm in the oligodendroglia of sporadic and COQ2 mutant multiple system atrophy. Acta Neuropathol. Commun. 2014, 2, 136. [Google Scholar] [CrossRef]
- Alfattal, R.; Alfarhan, M.; Algaith, A.M.; Albash, B.; Elshafie, R.M.; Alshammari, A.; Alahmad, A.; Dashti, F.; Alsafi, R.; Alsharhan, H. LYRM7-associated mitochondrial complex III deficiency with non-cavitating leukoencephalopathy and stroke-like episodes. Am. J. Med. Genet. A 2023. [Google Scholar] [CrossRef]
- Brockmann, K.; Bjornstad, A.; Dechent, P.; Korenke, C.G.; Smeitink, J.; Trijbels, J.M.F.; Athanassopoulos, S.; Villagran, R.; Skjeldal, O.H.; Wilichowski, E.; et al. Succinate in dystrophic white matter: A proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Ann. Neurol. 2002, 52, 38–46. [Google Scholar] [CrossRef]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef]
- Subramanian, K.; Jochem, A.; Le Vasseur, M.; Lewis, S.; Paulson, B.R.; Reddy, T.R.; Russell, J.D.; Coon, J.J.; Pagliarini, D.J.; Nunnari, J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER–mitochondria contacts. J. Cell Biol. 2019, 218, 1353–1369. [Google Scholar] [CrossRef]
- Watanabe, K.; Nozaki, S.; Goto, M.; Kaneko, K.-I.; Hayashinaka, E.; Irie, S.; Nishiyama, A.; Kasai, K.; Fujii, K.; Wada, Y.; et al. PET imaging of 11C-labeled coenzyme Q10: Comparison of biodistribution between [11C] ubiquinol-10 and [11C] ubiquinone-10. Biochem. Biophys. Res. Commun. 2019, 512, 611–615. [Google Scholar] [CrossRef]
- Prasuhn, J.; Göttlich, M.; Ebeling, B.; Bodemann, C.; Großer, S.; Wellach, I.; Reuther, K.; Hanssen, H.; Brüggemann, N. The cerebellar bioenergetic state predicts treatment response in COQ8A-related ataxia. Park. Relat. Disord. 2022, 99, 91–95. [Google Scholar] [CrossRef]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Díaz-Casado, M.E.; Sánchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. β-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9(R239X) mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta 2014, 1842, 1–6. [Google Scholar] [CrossRef]
- Herebian, D.; López, L.C.; Distelmaier, F. Bypassing human CoQ(10) deficiency. Mol. Genet. Metab. 2018, 123, 289–291. [Google Scholar] [CrossRef]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Rodenburg, R.J.; Mayatepek, E.; Distelmaier, F. 4-Hydroxybenzoic acid restores CoQ(10) biosynthesis in human COQ2 deficiency. Ann. Clin. Transl. Neurol. 2017, 4, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münch, J.; Prasuhn, J.; Laugwitz, L.; Fung, C.-W.; Chung, B.H.-Y.; Bellusci, M.; Mayatepek, E.; Klee, D.; Distelmaier, F. Neuroimaging in Primary Coenzyme-Q10-Deficiency Disorders. Antioxidants 2023, 12, 718. https://doi.org/10.3390/antiox12030718
Münch J, Prasuhn J, Laugwitz L, Fung C-W, Chung BH-Y, Bellusci M, Mayatepek E, Klee D, Distelmaier F. Neuroimaging in Primary Coenzyme-Q10-Deficiency Disorders. Antioxidants. 2023; 12(3):718. https://doi.org/10.3390/antiox12030718
Chicago/Turabian StyleMünch, Juliane, Jannik Prasuhn, Lucia Laugwitz, Cheuk-Wing Fung, Brian H.-Y. Chung, Marcello Bellusci, Ertan Mayatepek, Dirk Klee, and Felix Distelmaier. 2023. "Neuroimaging in Primary Coenzyme-Q10-Deficiency Disorders" Antioxidants 12, no. 3: 718. https://doi.org/10.3390/antiox12030718
APA StyleMünch, J., Prasuhn, J., Laugwitz, L., Fung, C. -W., Chung, B. H. -Y., Bellusci, M., Mayatepek, E., Klee, D., & Distelmaier, F. (2023). Neuroimaging in Primary Coenzyme-Q10-Deficiency Disorders. Antioxidants, 12(3), 718. https://doi.org/10.3390/antiox12030718