

Partitioning of Antioxidants in Edible Oil–Water Binary Systems and in Oil-in-Water Emulsions


Abstract
:1. Introduction
2. Physicochemical Basis of Partitioning: Balance of Intermolecular Interactions
3. Energetics of Partitioning: Thermodynamic Equations
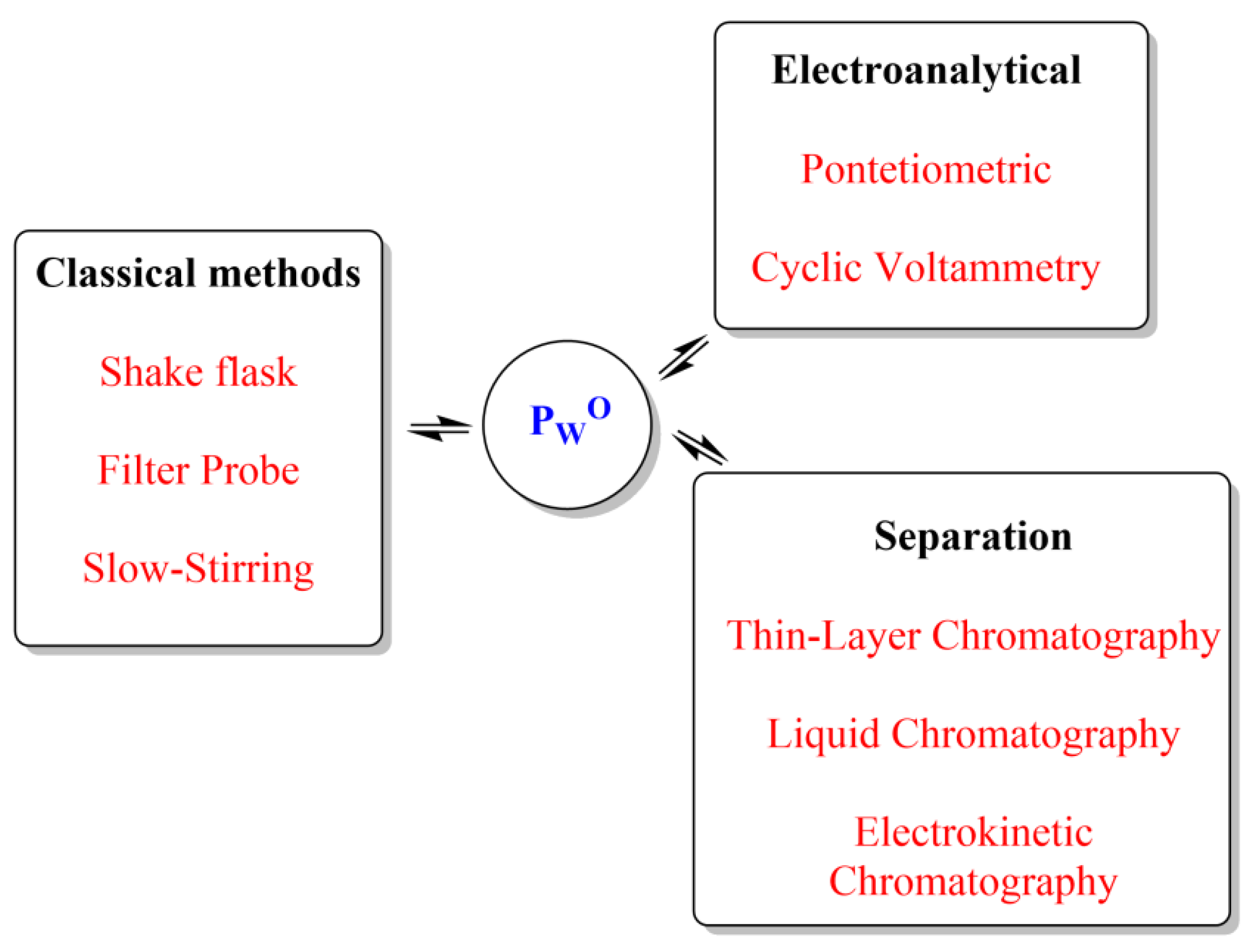
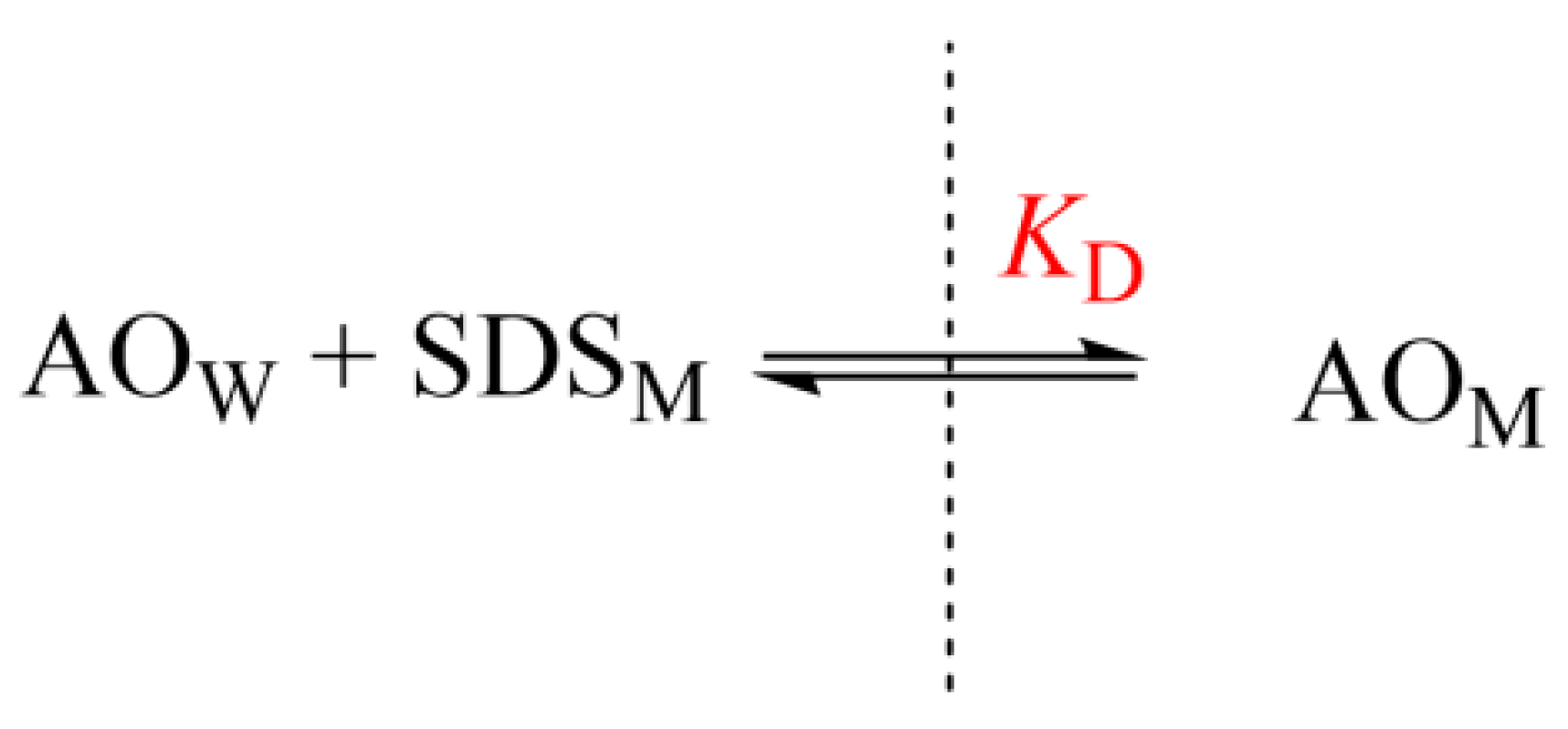
4. Methods to Measure Partition Constants
4.1. Experimental Methods
4.2. Computational Methods: Extrathermodynamic Approaches Based on Linear Free Energy Relationships (LFERs) to Predict and/or to Evaluate Partition Coefficients
Fragment-Based, Atom-Based, and Molecular Methods for Estimating Partition Constants
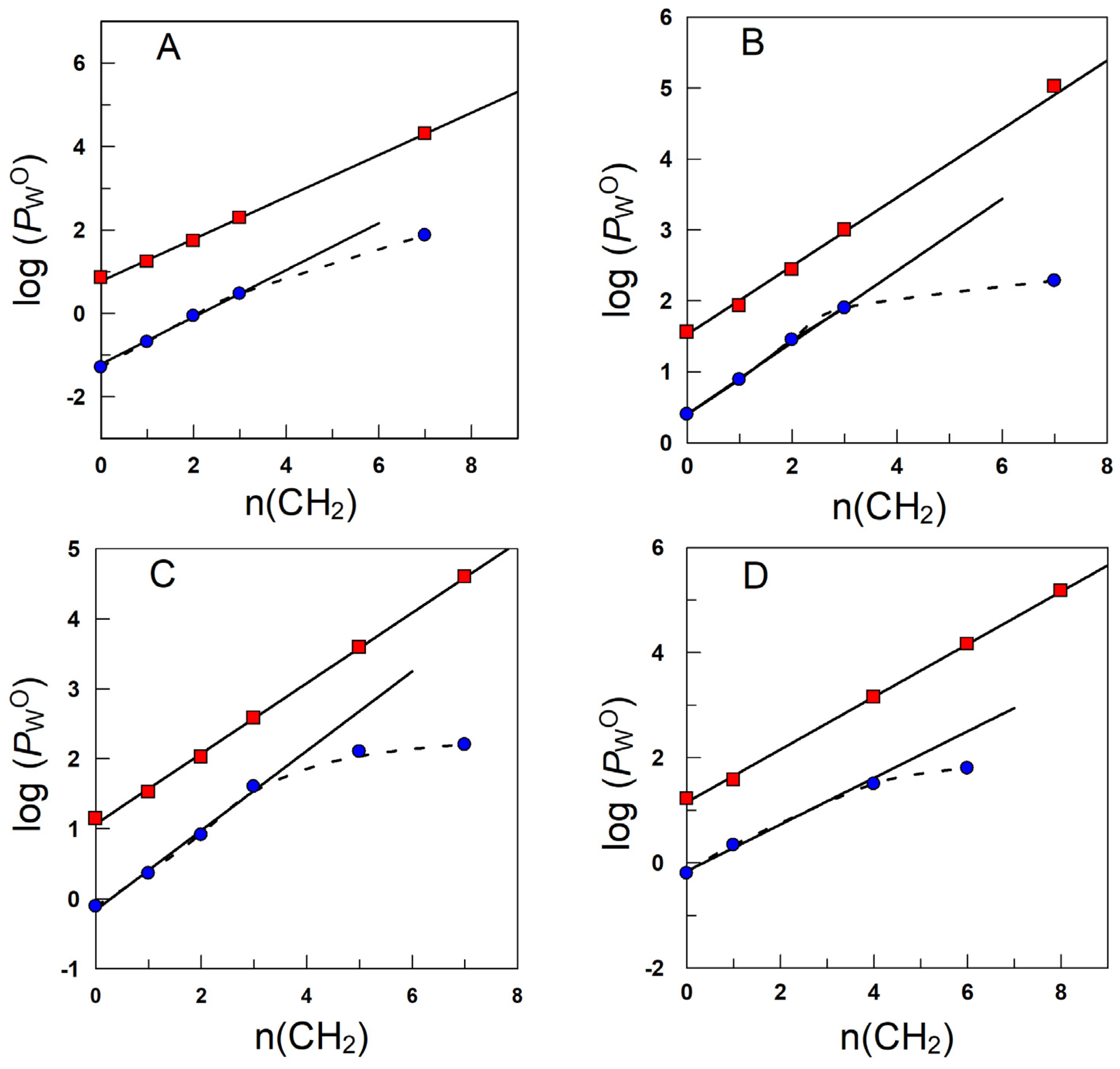
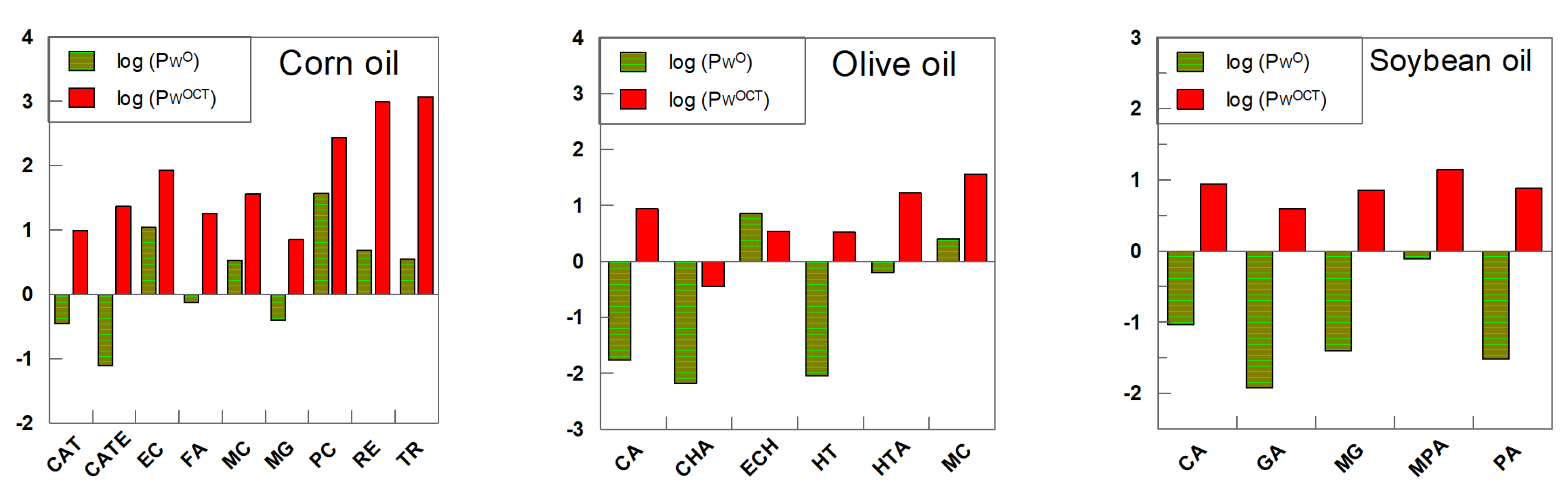
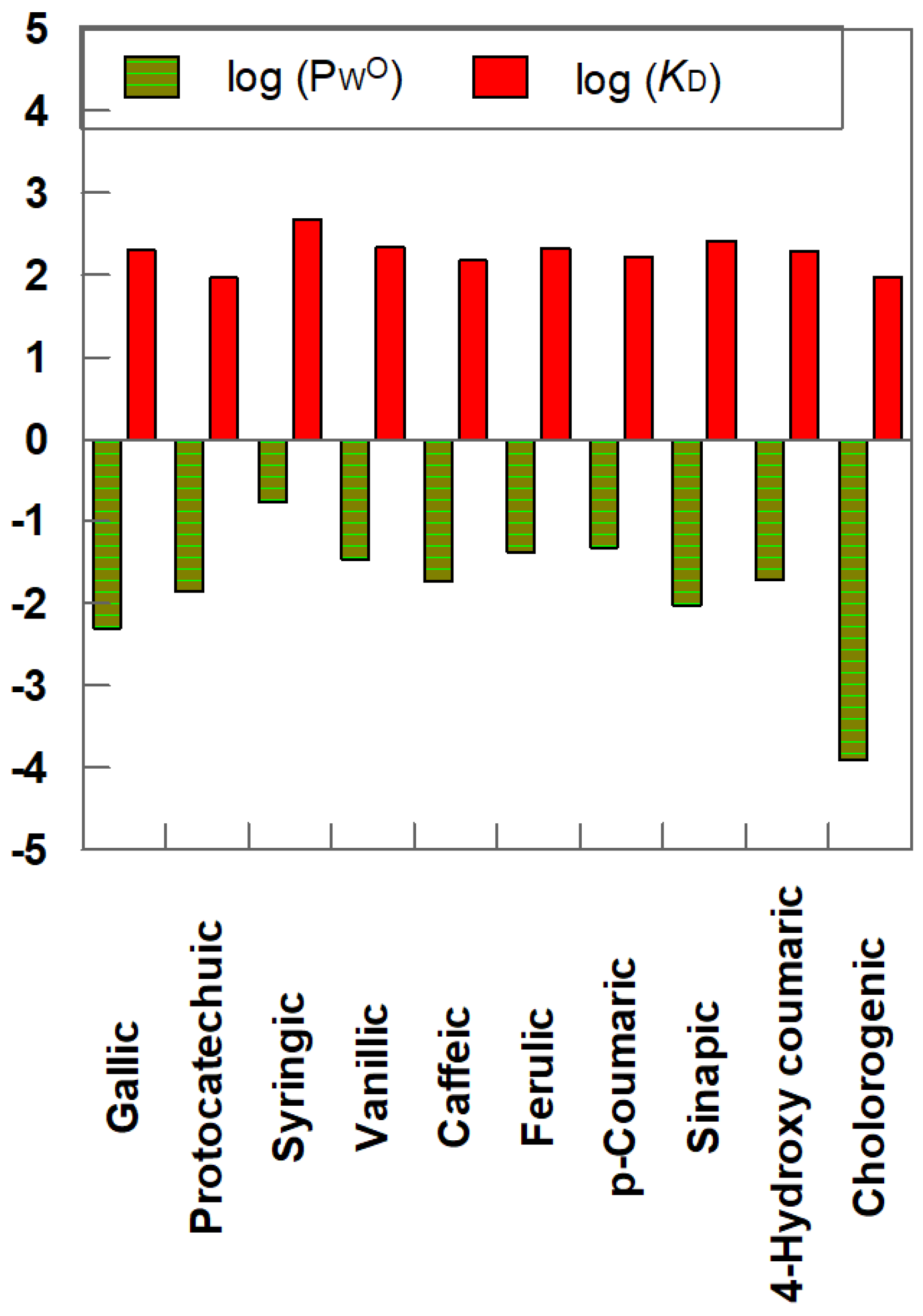
5. Partition Constants of Homologous Series of Antioxidants in Different Oils: Can the PWO Values Determined in Octanol–Water Systems Be Employed to Predict Those in Edible Oil–Water Systems?
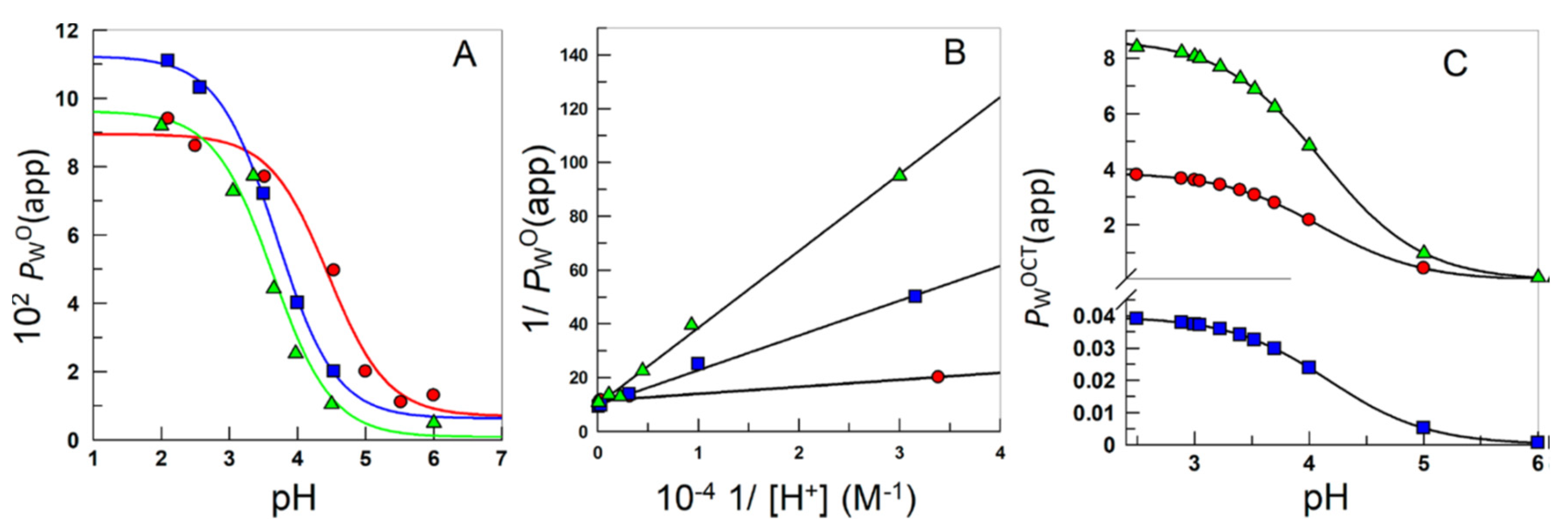
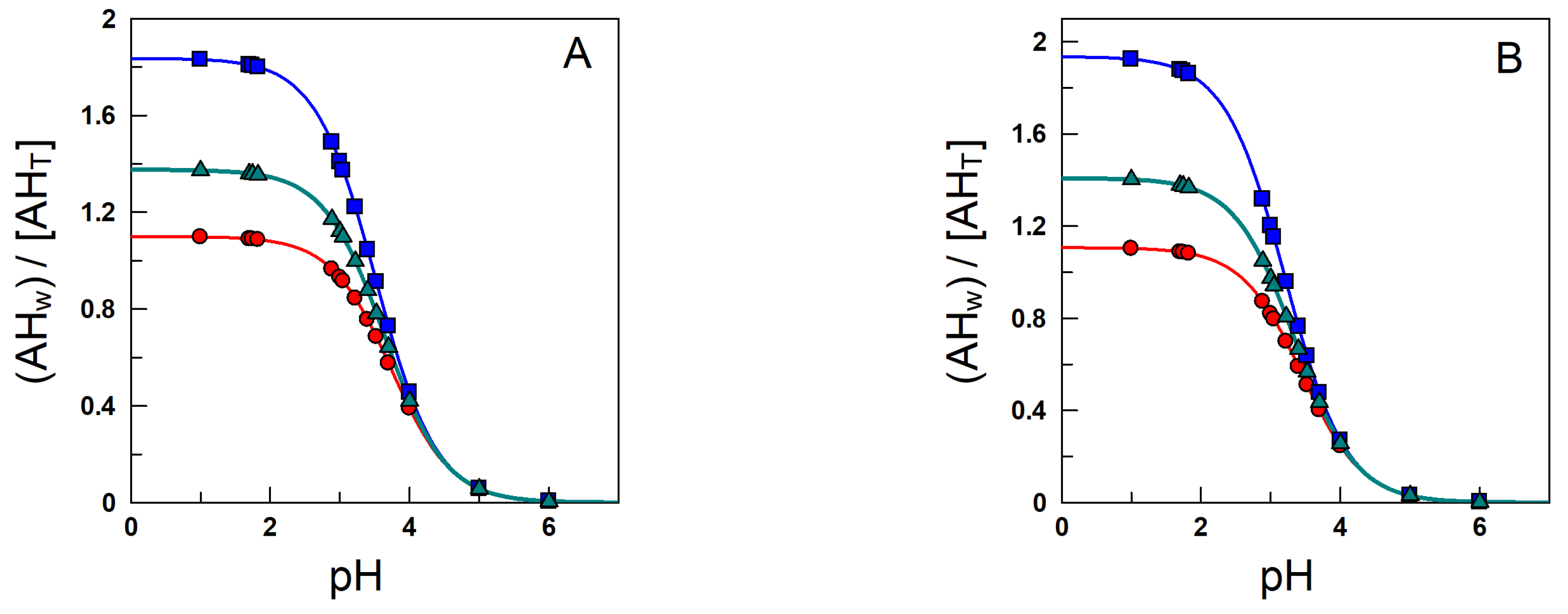
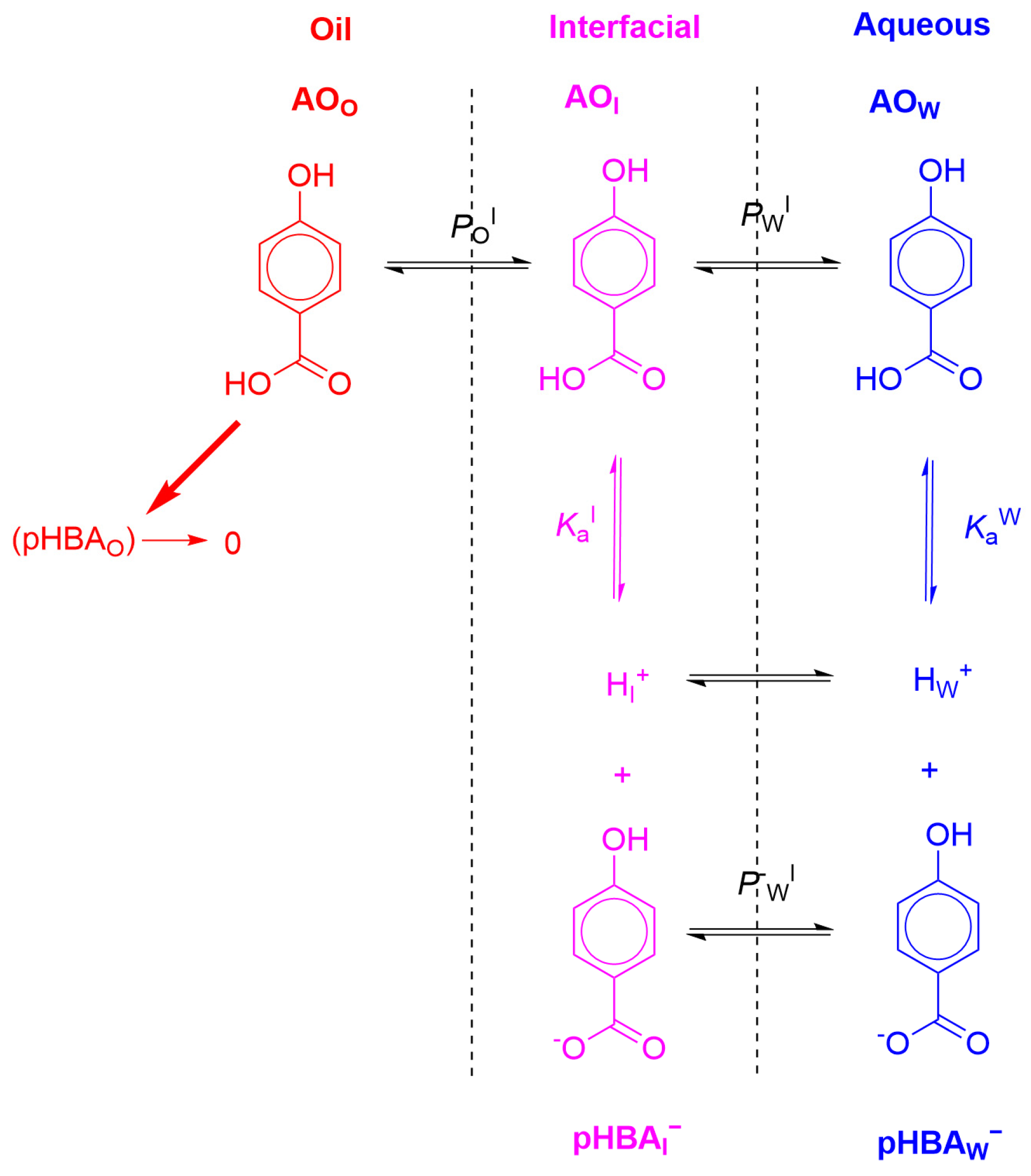
6. Effects of Acidity: Apparent Partition Coefficients of Ionizable Antioxidants
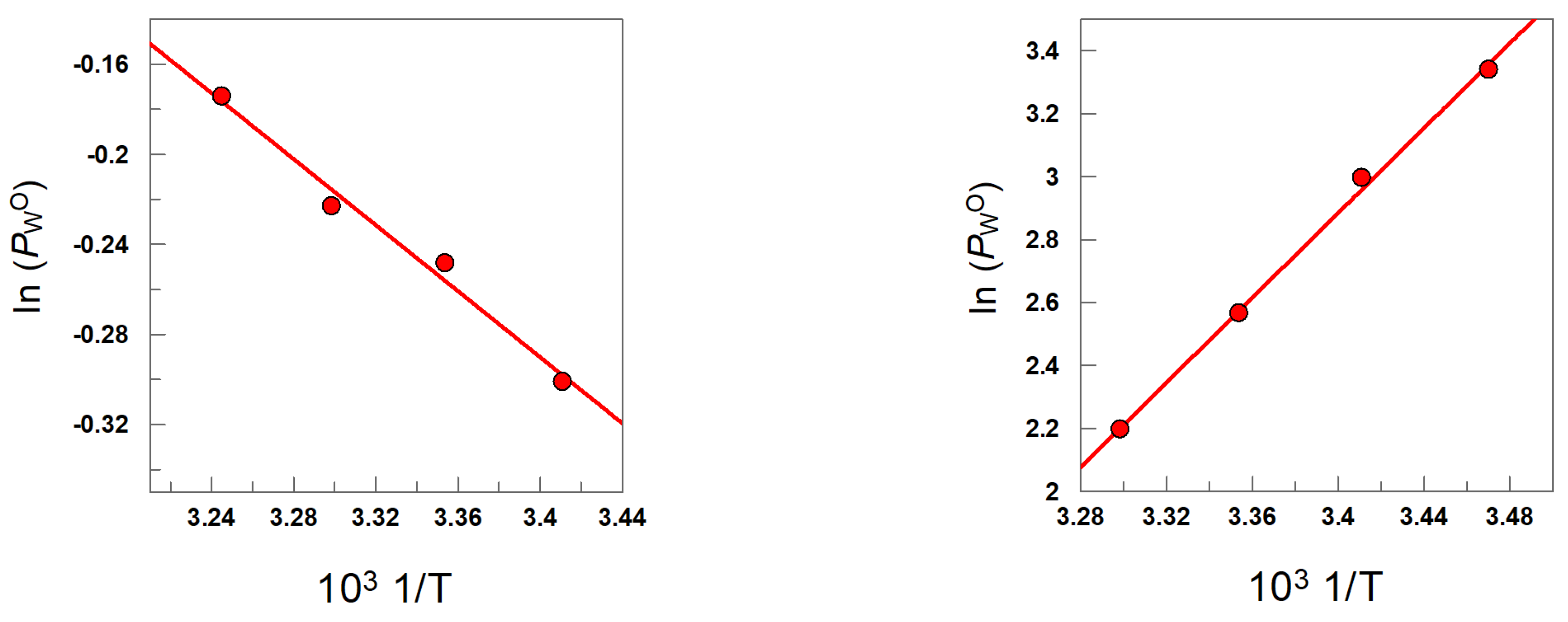
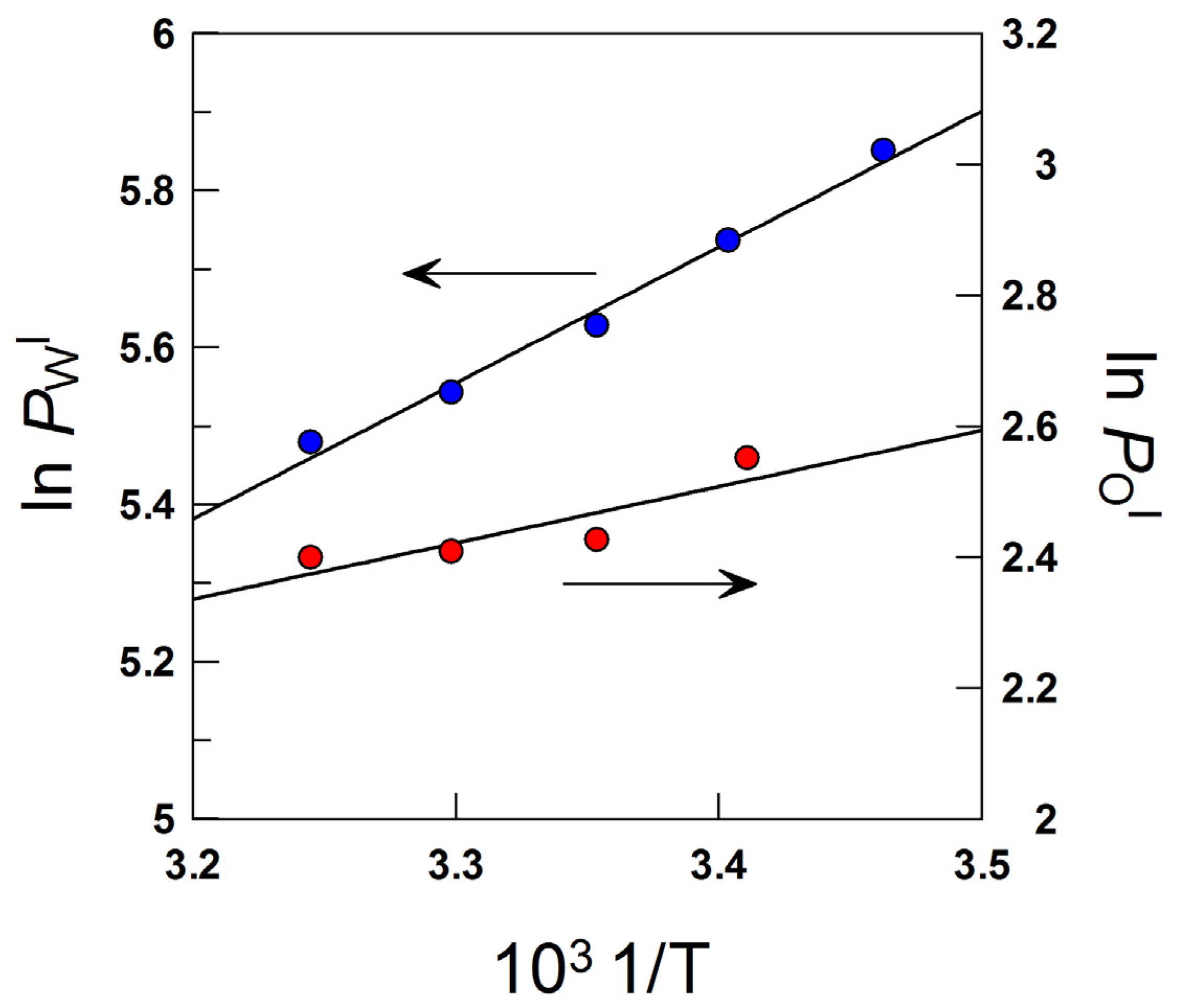
7. Effects of Temperature
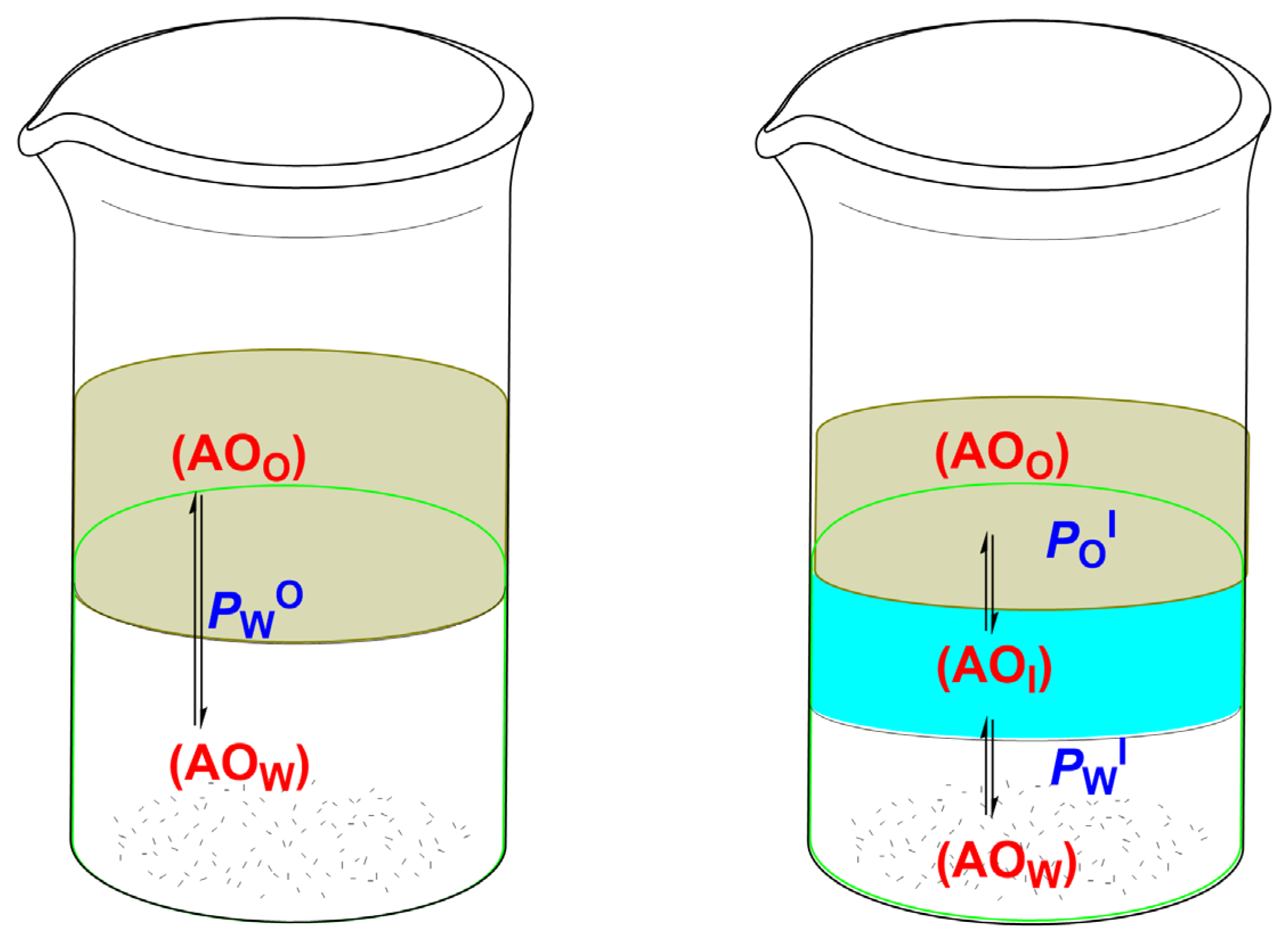
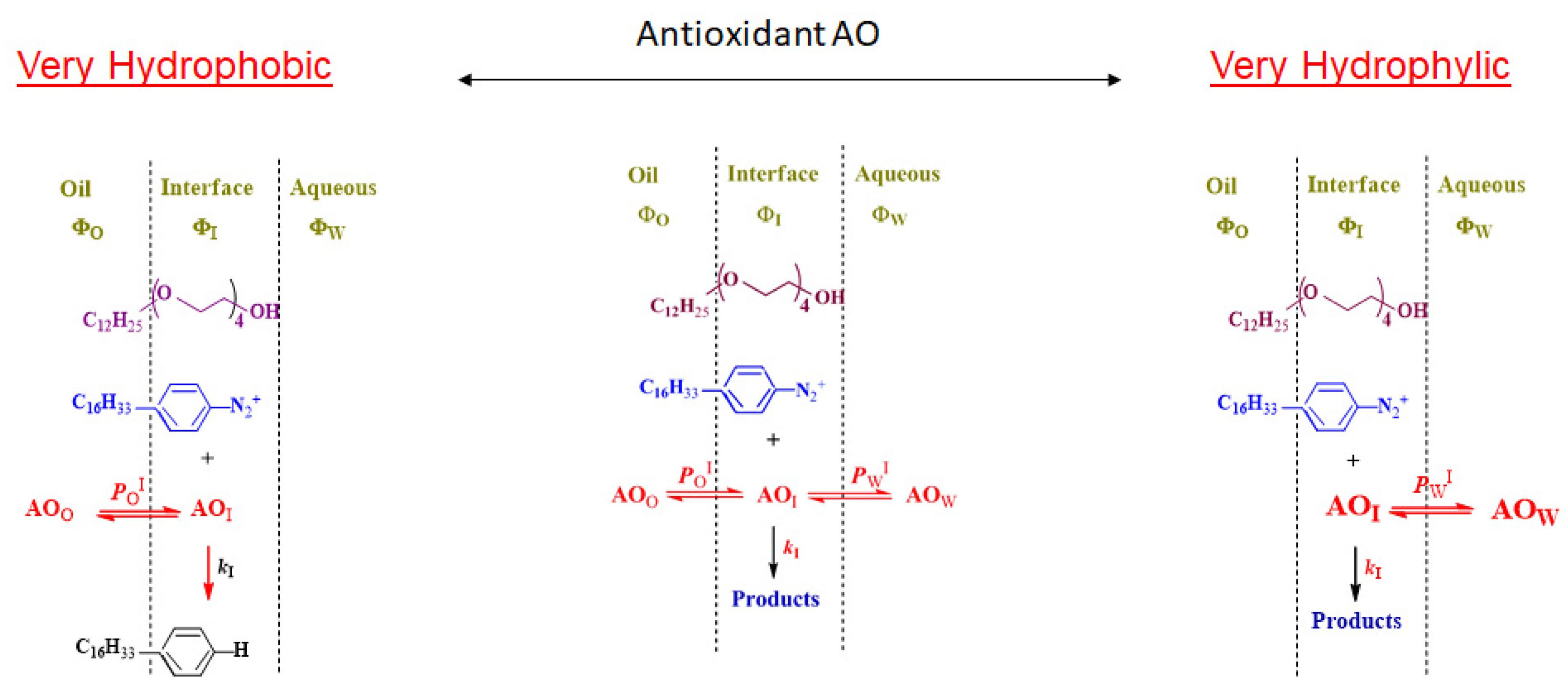
8. Partitioning in Emulsions
9. Final Remarks and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conde, E.; Díaz Reinoso, B.; González-Muñoz, M.J.; Moure, A.; Domínguez, H.; Parajó, J.C. Recovery and Concentration of Antioxidants from Industrial Effluents and from Processing Streams of Underutilized Vegetal Biomass. Food Public Health 2013, 3, 69–91. [Google Scholar] [CrossRef]
- Ben-Othman, S.; Jõudu, I.; Bhat, R. Bioactives from Agri-Food Wastes: Present Insights and Future Challenges. Molecules 2020, 25, 510. [Google Scholar] [CrossRef] [Green Version]
- Lourenço, S.C.; Moldão-Martins, M.; Alves, V.D. Antioxidants of Natural Plant Origins: From Sources to Food Industry Applications. Molecules 2019, 24, 4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.; Sezgin-Bayindir, Z.; Losada-Barreiro, S.; Paiva-Martins, F.; Saso, L.; Bravo-Díaz, C. Polyphenols as Antioxidants for Extending Food Shelf-Life and in the Prevention of Health Diseases: Encapsulation and Interfacial Phenomena. Biomedicines 2021, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Sezgin-Bayindir, Z.; Losada-Barreiro, S.; Paiva-Martins, F.; Saso, L.; Bravo-Díaz, C. Plant Antioxidants in Food Emulsions. In Some New Aspects of Colloidal Systems in Foods; Milani, J.M., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Jideani, A.I.O.; Silungwe, H.; Takalani, T.; Omolola, A.O.; Udeh, H.O.; Anyasi, T.A. Antioxidant-rich natural fruit and vegetable products and human health. Int. J. Food Prop. 2021, 24, 41–67. [Google Scholar] [CrossRef]
- Chandra, P.; Sharma, R.K.; Arora, D.S. Antioxidant compounds from microbial sources: A review. Food Res. Int. 2020, 129, 108849. [Google Scholar] [CrossRef]
- Díaz-Reinoso, B.; González-López, N.; Moure, A.; Domínguez, H.; Parajó, J.C. Recovery of antioxidants from industrial waste liquors using membranes and polymeric resins. J. Food Eng. 2010, 96, 127–133. [Google Scholar] [CrossRef]
- Fierascu, R.C.; Fierascu, I.; Avramescu, S.M.; Sieniawska, E. Recovery of Natural Antioxidants from Agro-Industrial Side Streams through Advanced Extraction Techniques. Molecules 2019, 24, 4212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.F.; Zhang, X.; Zhang, X.; Sverko, E.; Smyth, S.A.; Li, Y.F. Diphenylamine Antioxidants in wastewater influent, effluent, biosolids and landfill leachate: Contribution to environmental releases. Water Res. 2021, 189, 116602. [Google Scholar] [CrossRef]
- Justino, C.I.; Pereira, R.; Freitas, A.C.; Rocha-Santos, T.A.; Panteleitchouk, T.S.; Duarte, A.C. Olive oil mill wastewaters before and after treatment: A critical review from the ecotoxicological point of view. Ecotoxicology 2012, 21, 615–629. [Google Scholar] [CrossRef]
- Moure, A.; Domínguez, H.; Parajó, J.C. Antioxidant properties of ultrafiltration-recovered soy protein fractions from industrial effluents and their hydrolysates. Process Biochem. 2006, 41, 447–456. [Google Scholar] [CrossRef]
- Melini, V.; Melini, F.; Luziatelli, F.; Ruzzi, M. Functional Ingredients from Agri-Food Waste: Effect of Inclusion Thereof on Phenolic Compound Content and Bioaccessibility in Bakery Products. Antioxidants 2020, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Stromsnes, K.; Lagzdina, R.; Olaso-Gonzalez, G.; Gimeno-Mallench, L.; Gambini, J. Pharmacological Properties of Polyphenols: Bioavailability, Mechanisms of Action, and Biological Effects in In Vitro Studies, Animal Models, and Humans. Biomedicines 2021, 9, 1074. [Google Scholar] [CrossRef]
- Schaich, K.M. Lipid Antioxidants: More than Just Lipid Radical Quenchers. In Lipid Oxidation in Food and Biological Systems: A Physical Chemistry Perspective; Bravo-Diaz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 151–184. [Google Scholar] [CrossRef]
- Jimenez-Alvarez, D.; Giuffrida, F.; Golay, P.A.; Cotting, C.; Lardeau, A.; Keely, B.J. Antioxidant activity of oregano, parsley, and olive mill wastewaters in bulk oils and oil-in-water emulsions enriched in fish oil. J. Agric. Food Chem. 2008, 56, 7151–7159. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, Y.; Bai, R.; Zhang, X.; Yuan, L.; Liu, J. Preparation of pH-sensitive and antioxidant packaging films based on κ-carrageenan and mulberry polyphenolic extract. Int. J. Biol. Macromol. 2019, 134, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Barba, F.J.; Gómez, B.; Putnik, P.; Bursać Kovačević, D.; Pateiro, M.; Santos, E.M.; Lorenzo, J.M. Active packaging films with natural antioxidants to be used in meat industry: A review. Food Res. Int. 2018, 113, 93–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, B.; Lu, F.; Wang, L.; Ding, Y.; Kang, X. Plant-derived antioxidants incorporated into active packaging intended for vegetables and fatty animal products: A review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2021, 38, 1237–1248. [Google Scholar] [CrossRef]
- De Leonardis, A.; Macciola, V.; Lembo, G.; Aretini, A.; Nag, A. Studies on oxidative stabilisation of lard by natural antioxidants recovered from olive-oil mill wastewater. Food Chem. 2007, 100, 998–1004. [Google Scholar] [CrossRef]
- Leo, A.J. Octanol/Water Partition Coefficients. In Encyclopedia of Computational Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Berthod, A.; Carda-Bosch, S. Determination of liquid-liquid partition coefficients by separation methods. J. Chromatog. A 2004, 1037, 3–14. [Google Scholar] [CrossRef]
- Amézqueta, S.; Subirats, X.; Fuguet, E.; Rosés, M.; Ràfols, C. Chapter 6—Octanol-Water Partition Constant. In Liquid-Phase Extraction; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 183–208. [Google Scholar] [CrossRef]
- Loureiro, D.R.P.; Soares, J.X.; Lopes, D.; Macedo, T.; Yordanova, D.; Jakobtorweihen, S.; Nunes, C.; Reis, S.; Pinto, M.M.M.; Afonso, C.M.M. Accessing lipophilicity of drugs with biomimetic models: A comparative study using liposomes and micelles. Eur. J. Pharm. Sci. 2018, 115, 369–380. [Google Scholar] [CrossRef]
- Jakobtorweihen, S.; Zuniga, A.C.; Ingram, T.; Gerlach, T.; Keil, F.J.; Smirnova, I. Predicting solute partitioning in lipid bilayers: Free energies and partition coefficients from molecular dynamics simulations and COSMOmic. J. Chem. Phys. 2014, 141, 045102. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, S.V.; Ol’khovich, M.V.; Sharapova, A.V.; Proshin, A.N.; Perlovich, G.L. Partition coefficients and thermodynamics of transfer of novel drug-like spiro-derivatives in model biological solutions. J. Chem. Thermodyn. 2013, 61, 11–17. [Google Scholar] [CrossRef]
- Sangster, J. Octanol-Water Partition coefficients, Fundamentals and Physcial Chemistry; J. Wiley & Sons: Chichester, UK, 1997. [Google Scholar]
- Dini, I. Contribution of Nanoscience Research in Antioxidants Delivery Used in Nutricosmetic Sector. Antioxidants 2022, 11, 563. [Google Scholar] [CrossRef]
- Sezgin-Bayindir, Z.; Losada-Barreiro, S.; Bravo-Díaz, C.; Sova, M.; Kristl, J.; Saso, L. Nanotechnology-Based Drug Delivery to Improve the Therapeutic Benefits of NRF2 Modulators in Cancer Therapy. Antioxidants 2021, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Losada-Barreiro, S.; Bravo-Díaz, C.; Paiva-Martins, F. Why encapsulate antioxidants in emulsion-based systems, where they are located, and how location affects their efficiency. In Emulsion-Based Encapsulation of Antioxidants; Aboudzadeh, M.A., Ed.; Springer Nature: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- García-Pérez, P.; Losada-Barreiro, S.; Gallego, P.P.; Bravo-Díaz, C. Cyclodextrin-Elicited Bryophyllum Suspension Cultured Cells: Enhancement of the Production of Bioactive Compounds. Int. J. Mol. Sci. 2019, 20, 5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Chapter 15—Oral controlled and sustained drug delivery systems: Concepts, advances, preclinical, and clinical status. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 567–626. [Google Scholar] [CrossRef]
- Chamberlin, A.C.; Levitt, D.G.; Cramer, C.J.; Truhlar, D.G. Modeling Free Energies of Solvation in Olive Oil. Mol. Pharm. 2008, 5, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Coe, E.L.; Coe, M.H. A hypothesis relating oil: Water partition coefficients and vapor pressures of nonelectrolytes to their penetration rates through biological membranes. J. Theor. Biol. 1965, 8, 327–343. [Google Scholar] [CrossRef]
- Chiou, C.T. Partition coefficients of organic compounds in lipid-water systems and correlations with fish bioconcentration factors. Environ. Sci. Technol. 1985, 19, 57–62. [Google Scholar] [CrossRef]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and Its Relationship with Passive Drug Permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef]
- Poulin, P.; Schoenlein, K.; Theil, F.P. Prediction of adipose tissue: Plasma partition coefficients for structurally unrelated drugs. J. Pharm. Sci. 2001, 90, 436–447. [Google Scholar] [CrossRef]
- Krinsky, N.I. Membrane antioxidants. Ann. N.Y. Acad. Sci. 1988, 551, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Palmer, D.S.; Llinàs, A.; Morao, I.; Day, G.M.; Goodman, J.M.; Glen, R.C.; Mitchell, J.B.O. Predicting Intrinsic Aqueous Solubility by a Thermodynamic Cycle. Mol. Pharm. 2008, 5, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Paiva-Martins, F.; Bravo-Díaz, C.; Losada-Barreiro, S. Control of Lipid Oxidation in Oil-in Water Emulsions: Effects of Antioxidant Partitioning and Surfactant Concentration. In Lipid Oxidation in Food and Biological Systems: A Physical Chemistry Perspective; Bravo-Diaz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 201–216. [Google Scholar] [CrossRef]
- Romsted, L.S.; Bravo-Díaz, C. Determining Antioxidant Distributions in Intact Emulsions by Kinetic Methods: Application of Pseudophase Models. In Lipid Oxidation in Food and Biological Systems: A Physical Chemistry Perspective; Bravo-Diaz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 31–48. [Google Scholar] [CrossRef]
- Pérez-Palacios, T.; Estévez, M. Lipid Oxidation in Meat Systems: Updated Means of Detection and Innovative Antioxidant Strategies. In Lipid Oxidation in Food and Biological Systems: A Physical Chemistry Perspective; Bravo-Diaz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 93–111. [Google Scholar] [CrossRef]
- Costa, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Paiva-Martins, F. Effects of Emulsion Droplet Size on the Distribution and Efficiency of Antioxidants. In Lipid Oxidation in Food and Biological Systems: A Physical Chemistry Perspective; Bravo-Diaz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 217–235. [Google Scholar] [CrossRef]
- Bravo-Díaz, C. Advances in the control of lipid peroxidation in oil-in-water emulsions: Kinetic approaches. Crit. Rev. Food Sci. Nutr. 2022, 1, 1–33. [Google Scholar] [CrossRef]
- Wu, H.; Richards, M.P. Lipid oxidation and antioxidant delivery systems in muscle food. Compr. Rev. Food Sci. Food Saf. 2022, 21, 1275–1299. [Google Scholar] [CrossRef]
- Yang, W.; Yue, H.; Lu, G.; Wang, W.; Deng, Y.; Ma, G.; Wei, W. Advances in Delivering Oxidative Modulators for Disease Therapy. Research 2022, 2022, 9897464. [Google Scholar] [CrossRef]
- Nic, M.; Jirat, J.; Kosata, J.; Jenkins, A.; McNaught, A. Compendium of Chemical Terminology. Gold Book, IUPAC, (Web 2.0 Version); IUPAC: Research Triangle Park, NC, USA, 2019. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition Coefficients And Their Uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Chen, C.-S.; Lin, S.-T. Prediction of pH Effect on the Octanol–Water Partition Coefficient of Ionizable Pharmaceuticals. Ind. Eng. Chem. Res. 2016, 55, 9284–9294. [Google Scholar] [CrossRef]
- Bannan, C.C.; Calabró, G.; Kyu, D.Y.; Mobley, D.L. Calculating Partition Coefficients of Small Molecules in Octanol/Water and Cyclohexane/Water. J. Chem. Theory Comput. 2016, 12, 4015–4024. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity--shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef] [Green Version]
- Leckband, D.; Israelachvili, J. Intermolecular forces in biology. Q. Rev. Biophys. 2001, 34, 105–267. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Physical Chemistry; Oxford University Press: Oxford, UK, 2022. [Google Scholar]
- Israelachvili, J. Intermolecular and Surface Forces, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Leo, A.J.; Hansch, C. Linear free energy relations between partitioning solvent systems. J. Org. Chem. 1971, 36, 1539–1544. [Google Scholar] [CrossRef]
- Schwarzenbach, R.P.; Gschwend, P.M.; Imboden, D.M. Partitioning: Molecular Interactions and Thermodynamics. In Environmental Organic Chemistry; Schwarzenbach, R.P., Gschwend, P.M., Imboden, D.M., Eds.; J. Wiley & Sons: Hoboken, NJ, USA, 2002; pp. 57–96. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Hoelkman, D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Zamora, W.J.; Curutchet, C.; Campanera, J.M.; Luque, F.J. Prediction of pH-Dependent Hydrophobic Profiles of Small Molecules from Miertus–Scrocco–Tomasi Continuum Solvation Calculations. J. Phys. Chem. B 2017, 121, 9868–9880. [Google Scholar] [CrossRef]
- Kempińska, D.; Chmiel, T.; Kot-Wasik, A.; Mróz, A.; Mazerska, Z.; Namieśnik, J. State of the art and prospects of methods for determination of lipophilicity of chemical compounds. TrAC Trends Anal. Chem. 2019, 113, 54–73. [Google Scholar] [CrossRef]
- Montalbán, M.G.; Collado-González, M.; Trigo, R.M.; Fg, D.; Víllora, G. Experimental Measurements of Octanol-Water Partition Coefficients of Ionic Liquids. J. Adv. Chem. Eng. 2015, 5, 2. [Google Scholar] [CrossRef]
- Berthod, A.; García Álvarez-Coque, M.C. Micellar Liquid Chromatography; Chromatographic Science Series; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Liang, C.; Lian, H.-z. Recent advances in lipophilicity measurement by reversed-phase high-performance liquid chromatography. TrAC Trends Anal. Chem. 2015, 68, 28–36. [Google Scholar] [CrossRef]
- Kubik, Ł.; Struck-Lewicka, W.; Kaliszan, R.; Wiczling, P. Simultaneous determination of hydrophobicity and dissociation constant for a large set of compounds by gradient reverse phase high performance liquid chromatography–mass spectrometry technique. J. Chromatogr. A 2015, 1416, 31–37. [Google Scholar] [CrossRef]
- Kaliszan, R. QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef]
- Tetko, I.V.; Yan, A.; Gasteiger, J. Prediction of Physicochemical Properties of Compounds. In Applied Chemoinformatics; Wiley: Hoboken, NJ, USA, 2018; pp. 53–81. [Google Scholar] [CrossRef]
- Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Differential Partitioning of Bioantioxidants in Edible Oil–Water and Octanol–Water Systems: Linear Free Energy Relationships. J. Chem. Eng. Data 2018, 63, 2999–3007. [Google Scholar] [CrossRef]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Polyphenolic Antioxidants in Lipid Emulsions: Partitioning Effects and Interfacial Phenomena. Foods 2021, 10, 539. [Google Scholar]
- Molyneux, P. Octanol/water partition coefficients Kow: A critical examination of the value of the methylene group contribution to logKow for homologous series of organic compounds. Fluid Phase Equilibria 2014, 368, 120–141. [Google Scholar] [CrossRef]
- Cappelli, C.I.; Benfenati, E.; Cester, J. Evaluation of QSAR models for predicting the partition coefficient (logP) of chemicals under the REACH regulation. Environ. Res. 2015, 143, 26–32. [Google Scholar] [CrossRef]
- Klamt, A.; Eckert, F.; Arlt, W. COSMO-RS: An alternative to simulation for calculating thermodynamic properties of liquid mixtures. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannhold, R.; Rekker, R.F.; Sonntag, C.; ter Laak, A.M.; Dross, K.; Polymeropoulos, E.E. Comparative Evaluation of the Predictive Power of Calculation Procedures for Molecular Lipophilicity. J. Pharm. Sci. 1995, 84, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Beezer, A.E.; Gooch, C.A.; Hunter, W.H.; Volpe, P.L.O. A thermodynamic analysis of the Collander equation and establishment of a reference solvent for use in drug partitioning studies. J. Pharm. Pharmacol. 1987, 39, 774–779. [Google Scholar] [CrossRef]
- Molinspiration. Available online: https://www.molinspiration.com/ (accessed on 23 February 2023).
- Goss, K.-U.; Schwarzenbach, R.P. Linear Free Energy Relationships Used To Evaluate Equilibrium Partitioning of Organic Compounds. Environ. Sci. Technol. 2001, 35, 1–9. [Google Scholar] [CrossRef]
- Farooq, S.; Abdullah; Zhang, H.; Weiss, J. A comprehensive review on polarity, partitioning, and interactions of phenolic antioxidants at oil–water interface of food emulsions. Nat. Antioxid. Sources Compd. 2021, 20, 4250–4277. [Google Scholar] [CrossRef]
- Váňová, J.; Liimatta, L.J.; Česla, P.; Wiedmer, S.K. Determination of distribution constants of antioxidants by electrokinetic chromatography. Cogent Chem. 2017, 3, 1385173. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Bravo-Díaz, C.; Romsted, L.S. Distributions of phenolic acid antioxidants between the interfacial and aqueous regions of corn oil emulsions: Effects of pH and emulsifier concentration. Eur. J. Lipid Sci. Technol. 2015, 117, 1801–1813. [Google Scholar]
- Dean, J.A. Lange´s Handbook of Chemistry; McGraw-Hill, Inc.: New York, NY, USA, 1992. [Google Scholar]
- Fazary, A.E.; Ju, Y. Nonaqueous Solution Studies on the Protonation Equilibria of some Phenolic Acids. J. Solut. Chem. 2008, 37, 1305–1319. [Google Scholar] [CrossRef]
- Ozkorucuklu, S.P.; Beltrán, J.L.; Fonrodona, G.; Barrón, D.; Alsancak, G.; Barbosa, J. Determination of Dissociation Constants of Some Hydroxylated Benzoic and Cinnamic Acids in Water from Mobility and Spectroscopic Data Obtained by CE-DAD. J. Chem. Eng. Data 2009, 54, 807–811. [Google Scholar] [CrossRef]
- Arellano, J.B.; Li, H.; González-Pérez, S.; Gutiérrez, J.; Melø, T.B.; Vacha, F.; Naqvi, K.R. Trolox, a Water-Soluble Analogue of α-Tocopherol, Photoprotects the Surface-Exposed Regions of the Photosystem II Reaction Center in Vitro. Is This Physiologically Relevant? Biochem. 2011, 50, 8291–8301. [Google Scholar]
- Raimúndez-Rodríguez, E.A.; Losada-Barreiro, S.; Bravo-Díaz, C. Enhancing the fraction of antioxidants at the interfaces of oil-in-water emulsions: A kinetic and thermodynamic analysis of their partitioning. J. Colloid Interface Sci. 2019, 555, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Pastoriza-Gallego, M.J.; Sánchez-Paz, V.; Losada-Barreiro, S.; Bravo-Diaz, C.; Gunaseelan, K.; Romsted, L.S. Effects of Temperature and Emulsifier Concentration on α-Tocopherol Distribution in a Stirred, Fluid, Emulsion. Thermodynamics of α-Tocopherol transfer between the Oil and Interfacial Regions. Langmuir 2009, 25, 2646–2653. [Google Scholar] [PubMed]
- McClements, D.J. Food Emulsions, Principles, Practices and Techniques; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Dalgleish, D.G. Food emulsions—Their structures and structure-forming properties. Food Hydrocoll. 2006, 20, 415–422. [Google Scholar] [CrossRef]
- Dar, A.A.; Bravo-Diaz, C.; Nazir, N.; Romsted, L.S. Chemical kinetic and chemical trapping methods: Unique approaches for determining respectively the antioxidant distributions and interfacial molarities of water, counter-anions, and other weakly basic nucleophiles in association colloids. Curr. Opin. Colloid Interface Sci. 2017, 32, 84–93. [Google Scholar] [CrossRef]
- Bravo-Díaz, C.; Romsted, L.S.; Liu, C.; Losada-Barreiro, S.; Pastoriza-Gallego, M.J.; Gao, X.; Gu, Q.; Krishnan, G.; Sánchez-Paz, V.; Zhang, Y.; et al. To Model Chemical Reactivity in Heterogeneous Emulsions, Think Homogeneous Microemulsions. Langmuir 2015, 31, 8961–8979. [Google Scholar] [CrossRef] [Green Version]
- Romsted, L.S.; Bravo-Díaz, C. Modelling chemical reactivity in emulsions. Curr. Opin. Colloid Interface Sci. 2013, 18, 3–14. [Google Scholar]
- Losada-Barreiro, S.; Sánchez-Paz, V.; Bravo-Díaz, C. Transfer of antioxidants at the interfaces of model food emulsions: Distributions and thermodynamic parameters. Org. Biomol. Chem. 2008, 6, 4004–4011. [Google Scholar]
- Almeida, J.; Losada-Barreiro, S.; Costa, M.; Paiva-Martins, F.; Bravo-Díaz, C.; Romsted, L.S. Interfacial Concentrations of Hydroxytyrosol and Its Lipophilic Esters in Intact Olive Oil-in-Water Emulsions: Effects of Antioxidant Hydrophobicity, Surfactant Concentration, and the Oil-to-Water Ratio on the Oxidative Stability of the Emulsions. J. Agric. Food Chem. 2016, 64, 5274–5283. [Google Scholar]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C.; Romsted, L.S. A direct correlation between the antioxidant efficiencies of caffeic acid and its alkyl esters and their concentrations in the interfacial region of olive oil emulsions. The pseudophase model interpretation of the ‘‘cut-off’’ effect. Food Chem. 2015, 175, 233–242. [Google Scholar]
- Costa, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Monteiro, L.S.; Paiva-Martins, F. Interfacial Concentrations of Hydroxytyrosol Derivatives in Fish Oil-in-Water Emulsions and Nanoemulsions and Its Influence on Their Lipid Oxidation: Droplet Size Effects. Foods 2020, 9, 1897. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Effects of droplet size on the interfacial concentrations of antioxidants in fish and olive oil-in-water emulsions and nanoemulsions and on their oxidative stability. J. Colloid Interface Sci. 2020, 562, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Vicente, A.A.; Monteiro, L.S.; Paiva-Martins, F. Influence of AO chain length, droplet size and oil to water ratio on the distribution and on the activity of gallates in fish oil-in-water emulsified systems: Emulsion and nanoemulsion comparison. Food Chem. 2020, 310, 125716. [Google Scholar] [CrossRef] [Green Version]
- Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Enhancement of the antioxidant efficiency of gallic acid derivatives in intact fish oil-in-water emulsions through optimization of their interfacial concentrations. Food Funct. 2018, 9, 4429–4442. [Google Scholar]
- Martinez-Aranda, N.; Losada-Barreiro, N.; Bravo-Díaz, C.; Romsted, L.S. Influence of Temperature on the Distribution of Catechin in Corn oil-in-water Emulsions and some Relevant Thermodynamic Parameters. Food Biophys. 2014, 9, 380–388. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Bravo-Díaz, C.; Costa, M.; Paiva-Martins, F. Distribution of catechol in emulsions. J. Phys. Org. Chem. 2013, in press. [Google Scholar] [CrossRef]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Diaz, C. Optimizing the efficiency of antioxidants in emulsions by lipophilization: Tuning interfacial concentrations. RSC Adv. 2016, 6, 91483–91493. [Google Scholar]
- Losada-Barreiro, S.; Bravo Díaz, C.; Paiva Martins, F.; Romsted, L.S. Maxima in antioxidant distributions and efficiencies with increasing hydrophobicity of gallic acid and its alkyl esters. The pseudophase model interpretation of the “Cut-off effect. J. Agric. Food Chem. 2013, 61, 6533–6543. [Google Scholar]
- Narkiewicz-Michalek, J.; Szymula, M.; Losada-Barreiro, S.; Bravo-Diaz, C. Concentration of resveratrol at the oil–water interface of corn oil-in-water emulsions. Adsorption 2019, 25, 903–911. [Google Scholar] [CrossRef]
- Lisete-Torres, P.; Losada-Barreiro, S.; Albuquerque, H.; Sánchez-Paz, V.; Paiva-Martins, F.; Bravo-Díaz, C. Distribution of hydroxytyrosol and hydroxytyrosol acetate in olive oil emulsions and their antioxidant efficiency. J. Agric. Food Chem. 2012, 60, 7318–7325. [Google Scholar] [PubMed]
- Galan, A.; Losada-Barreiro, S.; Bravo-Díaz, C. A Physicochemical Study of the Effects of Acidity on the Distribution and Antioxidant Efficiency of Trolox in Olive Oil-in-Water Emulsions. ChemPhysChem 2016, 17, 296–304. [Google Scholar]
- Meireles, M.; Losada-Barreiro, S.; Costa, M.; Paiva-Martins, F.; Bravo-Díaz, C.; Monteiro, L.S. Control of antioxidant efficiency of chlorogenates in emulsions: Modulation of antioxidant interfacial concentrations. J. Sci. Food Agric. 2019, 99, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Mitrus, O.; Żuraw, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Paiva-Martins, F. Targeting Antioxidants to Interfaces: Control of the Oxidative Stability of Lipid-Based Emulsions. J. Agric. Food Chem. 2019, 67, 3266–3274. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Physical evidence that the variations in the efficiency of homologous series of antioxidants in emulsions are a result of differences in their distribution. J. Sci. Food Agric. 2017, 97, 564–571. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Force | Energy (kJ/mol) | Interacting Species |
|---|---|---|
| Ionic bond | 300–600 | Ions/Ions |
| Hydrogen bonding | 20–40 | Polar molecules containing N-H, O-H, or F-H, the link is a shared H atom |
| Ion–Dipole | 10–20 | Ion/polar molecule |
| Dipole–Dipole (Keeson) | 1–5 | polar molecule/polar molecule |
| Dipole–Induced dipole (Debye) | 2–10 | Stationary polar molecules/all types of molecules |
| Induced dipole–Induced dipole (London dispersion forces) | <2 | All types of molecules/all types of molecules |
| Experimental | |||
|---|---|---|---|
| Method | Advantage | Weakness | Applicability |
| Shake-flask | Most realistic, reliable, low experimental demand | Time consuming, large amounts of mutually saturated solvents required, emulsification may be a problem | Molecules of moderate hydrophobicity. Usually not recommended for very hydrophobic or hydrophilic compounds and when the tested substance dissociates |
| Slow-stirring methods | Avoid formation of emulsions | Time consuming, requires large amounts of solvent and product | Similar to the shake-flask method |
| Reverse-phase chromatography | Rapid, does not require large amounts of product nor solvent | Poor reproducibility because of different retention mechanisms, requires HPLC instrumentation | |
| Micellar electrokinetic chromatography | Good agreement with shake-flask method | Applicable to ionic substances | |
| Filter probe methods | Rapid | Expensive lab set-up, time consuming | |
| Software packages | |||
| Name | Company | Freeware | Comments |
| ACD/logD | Advanced Chemistry Development (www.acdlabs.com) | No | Fragment-based |
| ADMET predictor | Simulation Plis Inc. (www.simulationsplus.com) | No | Neural network |
| AlogP | Virtual Computation Chemistry Laboratory (www.vcclab.org) | Yes | Neural network |
| Hyperchem | Hypercube Inc. (www.hypercube.com) | No | Atom-additive method |
| MolInspiration | Molinspiration Cheminformatics, (https://www.mlinspiration.com) | Yes | Fragment based |
| SPARC | Univeristy of Georgia (http://www.ibmlc2.chem.uga.edu/sparc/) | Yes | Allows calculations under different ionic strength conditions |
| Partition Constant/Coefficients Correlated | LFER |
|---|---|
| Octanol–water (oct–w)/aqueous solubility (sat) | |
| Organic carbon–water (oc–w)/octanol–water (oct–w) | |
| Lipid–water (lip–w)/octanol–water (oct–w) |
| ANTIOXIDANT | Log PWO | |||||
|---|---|---|---|---|---|---|
| Structure | -R | n(CH2) | Olive | Soybean | Corn | Octanol |
 | -CH3 (C1) | 0 | 0.40 | 0.45 | 0.52 | 1.56 |
| -CH2 CH3 (C2) | 1 | 0.89 | 0.92 | 1.04 | 1.93 | |
| -(CH2)2 CH3 (C3) | 2 | 1.45 | 1.48 | 1.58 | 2.44 | |
| -(CH2)7 CH3 (C8) | 7 | 2.23 | 2.25 | 2.81 | 5.02 | |
 | -CH3 (C1) | 0 | −1.30 | −1.40 | −0.94 | 0.85 |
| -CH2 CH3 (C2) | 1 | −0.70 | --- | −0.60 | 1.23 | |
| -(CH2)2 CH3 (C3) | 2 | −0.07 | −0.07 | 0.10 | 1.73 | |
| -(CH2)3 CH3 (C4) | 3 | 0.48 | 0.51 | 0.60 | 2.29 | |
| -(CH2)7 CH3 (C8) | 7 | 1.88 | 2.29 | 4.31 | ||
 | CH3 (C2) | 0 | −0.20 | --- | 1.22 | |
| CH2 CH3 (C3) | 1 | 0.34 | --- | 1.58 | ||
| (CH2)4 CH3 (C6) | 4 | 1.50 | --- | 3.15 | ||
| (CH2)6 CH3 (C8) | 6 | 1.61 | --- | 4.16 | ||
 | -CH3 (C1) | 0 | −0.11 | −0.11 | 1.14 | |
| -CH2 CH3 (C2) | 1 | 0.27 | 0.36 | 1.52 | ||
| -(CH2)2 CH3 (C3) | 2 | 0.91 | 0.91 | 2.02 | ||
| -(CH2)3 CH3 (C4) | 3 | --- | 1.60 | 2.58 | ||
| -(CH2)5 CH3 (C6) | 5 | 1.72 | 1.71 | 3.59 | ||
| OIL | Antioxidants | Gallic | Caffeic | Protocatechuic | Hydroxytyrosol |
|---|---|---|---|---|---|
| Octanol (OCT) | aOCT | 0.77 ± 0.03 | 1.53 ± 0.05 | 1.06 ± 0.03 | 1.15 ± 0.04 |
| bOCT | 0.51 ± 0.01 | 0.49 ± 0.01 | 0.50 ± 0.01 | 0.50 ± 0.01 | |
| Olive (OL) | aOL | −1.15 ± 0.04 | 0.40 ± 0.03 | −0.18 ± 0.07 | −0.16 ± 0.02 |
| bOL | 0.50 ± 0.01 | 0.51 ± 0.01 | 0.55 ± 0.04 | 0.44 ± 0.02 | |
| Soybean (SO) | aSO | −1.18 ± 0.06 | 0.44 ± 0.03 | --- | --- |
| bSO | 0.58 ± 0.03 | 0.52 ± 0.03 | --- | --- | |
| Corn (CO) | aCO | −1.03 ± 0.10 | 0.52 ± 0.01 | --- | --- |
| bCO | 0.54 ± 0.06 | 0.53 ± 0.01 | --- | --- |
| Phenolic Acid | Molecular Structure | pKa(1) | pKa(2) | Ref |
|---|---|---|---|---|
| p-Coumaric |  | 4.37 | 9.20 | [80] |
| Caffeic |  | 3.94 4.30 | 8.47 8.51 | [80] [81] |
| Ferulic |  | 4.50 4.30 | 9.21 8.81 | [80] [81] |
| 3,4 Dihydroxy phenylacetic |  | 3.21 | 9.33 | [80] |
| Gallic acid |  | 4.10 4.11 | 8.38 8.47 | [80] [81] |
| Vanillic |  | 4.58 4.17 | 9.39 8.81 | [80] [81] |
| p-Hydroxybenzoic |  | 4.40 4.26 | 9.54 8.84 | [80] [81] |
| Trolox |  | 3.89 | 11.92 | [82] |
| Antioxidant | Surfactant | Oil/Water Ratio (v:v) | pH | PwI | POI | Reference |
|---|---|---|---|---|---|---|
| Fish oil | ||||||
| GA | TW80 | 4:6 | 3.7 | 118 | --- | [95] |
| GA | TW80 | 1:9 | 3.0 | 85 | --- | [96] |
| EG | TW80 | 4:6 | 3.7 | 233 | 706 | [95] |
| PG | TW80 | 1:9 | 3.0 | 154 | 101 | [96] |
| BG | TW80 | 4:6 | 3.7 | 559 | 253 | [95] |
| OG | TW80 | 4:6 | 3.7 | --- | 183 | [95] |
| LG | TW80 | 4:6 | 3.7 | --- | 142 | [95] |
| HT | TW80 | 1:9 | 3.7 | 34 | --- | [93] |
| EHT | TW80 | 1:9 | 3.7 | 207 | 115 | [93] |
| HET | TW80 | 1:9 | 3.7 | --- | 89 | [93] |
| OHT | TW80 | 1:9 | 3.7 | --- | 119 | [93] |
| LHT | TW80 | 1:9 | 3.7 | --- | 97 | [93] |
| HHT | TW80 | 1:9 | 3.7 | --- | 75 | [93] |
| Corn oil | ||||||
| CATE | TW20 | 4:6 | 2.1 | 368 | --- | [97] |
| CAT | TW20 | 4:6 | 3.7 | 57 | 170 | [98] |
| CA | TW20 | 4:6 | 3.7 | 268 | [99] | |
| PG | TW20 | 1:9 | 3.7 | 204 | 242 | [100] |
| OG | TW20 | 1:9 | 3.7 | --- | 29.8 | [100] |
| LG | TW20 | 3:7 | 3.7 | --- | 16.5 | [100] |
| RES | TW20 | 4:6 | 2.1 | 4076 | 860 | [101] |
| TOC | TW20 | 1:9 | 3.7 | --- | 11.3 | [90] |
| Olive oil | ||||||
| HT | TW20/SP80 | 1:9 | 3.7 | 120 | --- | [102] |
| HTA | TW20/SP80 | 1:9 | 3.6 | 204 | 331 | [102] |
| TR | TW20 | 4:6 | 2.2 | 5371 | 1773 | [103] |
| CGA | TW20 | 4:6 | 3.7 | 40 | --- | [104] |
| ECG | TW20 | 4:6 | 3.7 | 78 | --- | [104] |
| PCG | TW20 | 4:6 | 3.7 | 141 | --- | [104] |
| OCG | TW20 | 4:6 | 3.7 | --- | 111 | [104] |
| DCG | TW20 | 4:6 | 3.7 | --- | 124 | [104] |
| LCG | TW20 | 4:6 | 3.7 | --- | 159 | [104] |
| HCG | TW20 | 4:6 | 3.7 | --- | 89 | [104] |
| Soybean oil | ||||||
| GA | TW20 | 1:9 | 3.0 | 298 | [105] | |
| MG | TW20 | 1:9 | 3.0 | 329 | --- | [105] |
| PG | TW20 | 1:9 | 3.0 | 401 | 474 | [105] |
| BG | TW20 | 1:9 | 3.0 | 789 | 243 | [105] |
| OG | TW20 | 1:9 | 3.0 | 33 | --- | [105] |
| LG | TW20 | 1:9 | 3.0 | 23 | --- | [105] |
| CA | TW20 | 4:6 | 3.7 | 104 | --- | [106] |
| MC | TW20 | 4:6 | 3.7 | 445 | 150 | [106] |
| EC | TW20 | 4:6 | 3.7 | 1355 | 159 | [106] |
| PC | TW20 | 4:6 | 3.7 | 4727 | 164 | [106] |
| OC | TW20 | 4:6 | 3.7 | --- | 216 | [106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Partitioning of Antioxidants in Edible Oil–Water Binary Systems and in Oil-in-Water Emulsions. Antioxidants 2023, 12, 828. https://doi.org/10.3390/antiox12040828
Losada-Barreiro S, Paiva-Martins F, Bravo-Díaz C. Partitioning of Antioxidants in Edible Oil–Water Binary Systems and in Oil-in-Water Emulsions. Antioxidants. 2023; 12(4):828. https://doi.org/10.3390/antiox12040828
Chicago/Turabian StyleLosada-Barreiro, Sonia, Fátima Paiva-Martins, and Carlos Bravo-Díaz. 2023. "Partitioning of Antioxidants in Edible Oil–Water Binary Systems and in Oil-in-Water Emulsions" Antioxidants 12, no. 4: 828. https://doi.org/10.3390/antiox12040828
APA StyleLosada-Barreiro, S., Paiva-Martins, F., & Bravo-Díaz, C. (2023). Partitioning of Antioxidants in Edible Oil–Water Binary Systems and in Oil-in-Water Emulsions. Antioxidants, 12(4), 828. https://doi.org/10.3390/antiox12040828