
Novel Therapeutic Approaches to Liver Fibrosis Based on Targeting Oxidative Stress



{kind=link}
{kind=link}
Abstract
:1. Introduction
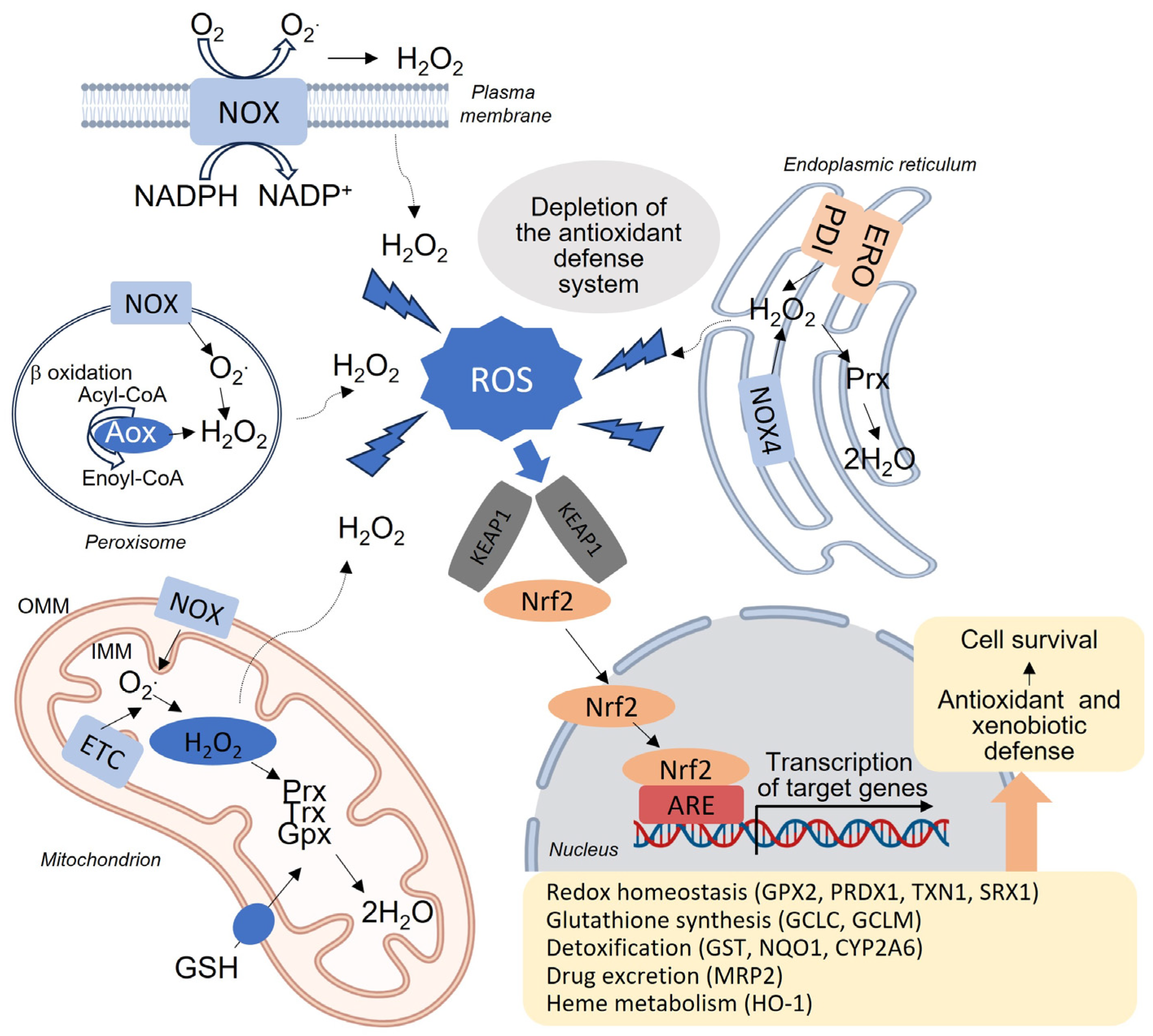
2. Mitochondria, Oxidative Stress and Liver Redox Homeostasis
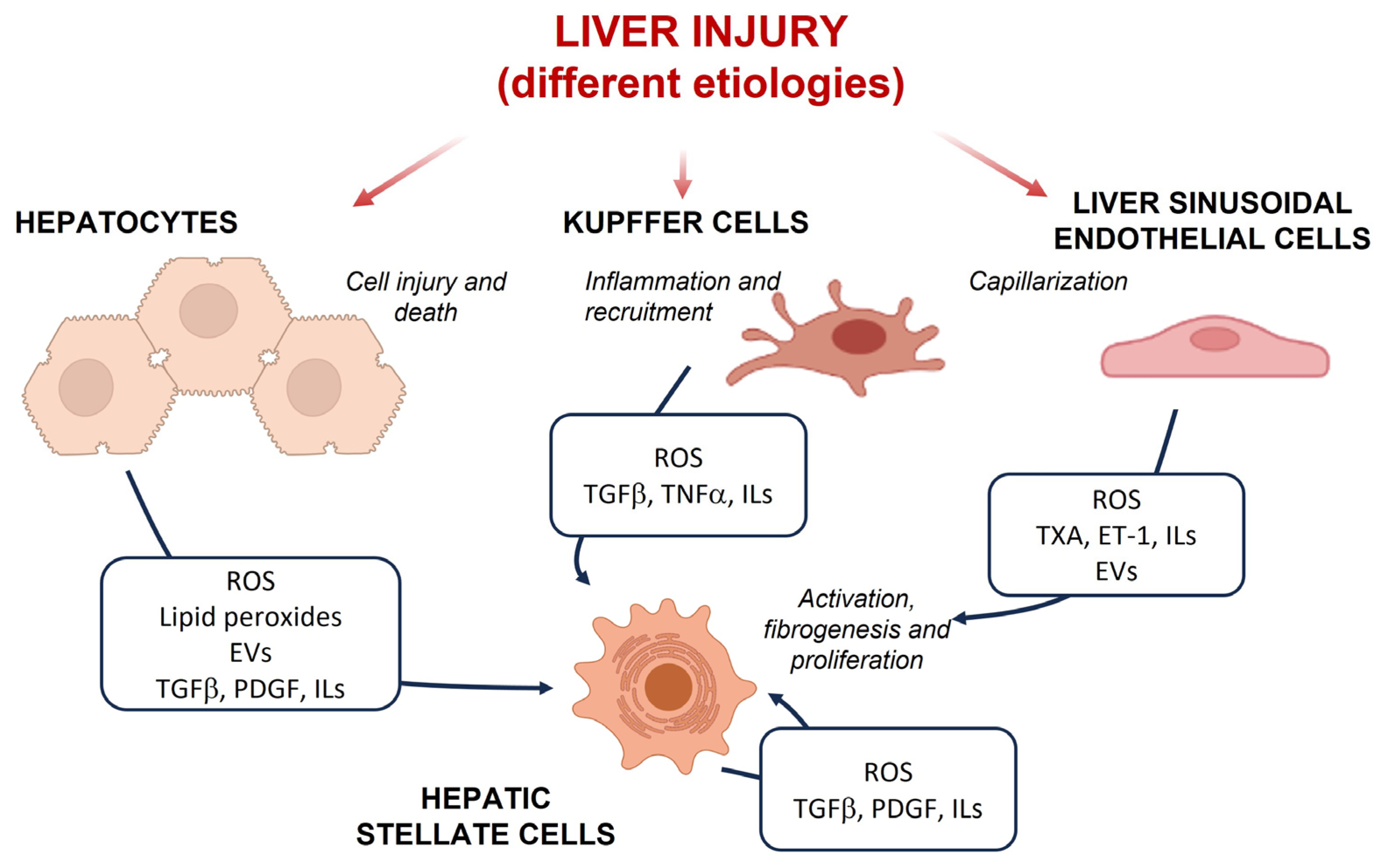
3. Oxidative Stress in Liver Cell Populations during CLD
3.1. Hepatocytes
3.2. Hepatic Stellate Cells
3.3. Macrophages
3.4. Liver Sinusoidal Endothelial Cells
4. Novel Therapeutic Approaches Targeting Oxidative Stress
4.1. Activation of Nrf2
4.2. NOX4/1 Inhibition
4.3. Peroxisome Proliferator-Activated Receptor γ (PPAR-γ) Activation
4.4. ER Stress and UPR
4.5. Mitochondrial Dysfunction and Mitochondrial Quality Control
4.6. Regulation of Cell Death: Ferroptosis and Pyroptosis
4.6.1. Ferroptosis
4.6.2. Pyroptosis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Swain, M.G.; Ramji, A.; Patel, K.; Sebastiani, G.; Shaheen, A.A.; Tam, E.; Marotta, P.; Elkhashab, M.; Bajaj, H.S.; Estes, C.; et al. Burden of nonalcoholic fatty liver disease in Canada, 2019–2030: A modelling study. CMAJ Open 2020, 8, E429–E436. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Shang, Z.; Gao, Y.; Wu, H.; Kong, X. Liver Regeneration in Chronic Liver Injuries: Basic and Clinical Applications Focusing on Macrophages and Natural Killer Cells. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 971–981. [Google Scholar] [CrossRef]
- Huang, R.; Zhang, X.; Gracia-Sancho, J.; Xie, W.F. Liver regeneration: Cellular origin and molecular mechanisms. Liver Int. 2022, 42, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 742931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascè, A.; Gariani, K.; Jornayvaz, F.R.; Szanto, I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef]
- Gill, R.; Tsung, A.; Billiar, T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic. Biol. Med. 2010, 48, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, S.; Kawanaka, M.; Kamei, A.; Izumi, A.; Nakata, K.; Niiyama, G.; Ikeda, H.; Hanano, S.; Suehiro, M.; Togawa, K.; et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid. Redox Signal. 2004, 6, 19–24. [Google Scholar] [CrossRef]
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; De Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: A pilot study. Oxidative Med. Cell. Longev. 2014, 2014, 169216. [Google Scholar] [CrossRef]
- Chung, F.L.; Nath, R.G.; Ocando, J.; Nishikawa, A.; Zhang, L. Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: Detection and potential sources. Cancer Res. 2000, 60, 1507–1511. [Google Scholar]
- Pamplona, R.; Costantini, D. Molecular and structural antioxidant defenses against oxidative stress in animals. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R843–R863. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kobayashi, Y.; Kawata, K.; Kawamura, K.; Sumiyoshi, S.; Noritake, H.; Watanabe, S.; Chida, T.; Souda, K.; Sakaguchi, T.; et al. Does hepatic oxidative stress enhance activation of nuclear factor-E2-related factor in patients with nonalcoholic steatohepatitis? Antioxid. Redox Signal. 2014, 20, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Ziros, P.G.; Zaravinos, A.; Iskrenova, R.P.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Hepatic gene expression profiling in Nrf2 knockout mice after long-term high-fat diet-induced obesity. Oxidative Med. Cell. Longev. 2013, 2013, 340731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.Y.; Elmasry, M.; Forootan, S.S.; Russomanno, G.; Bunday, T.M.; Zhang, F.; Brillant, N.; Starkey Lewis, P.J.; Aird, R.; Ricci, E.; et al. Pharmacological Activation of Nrf2 Enhances Functional Liver Regeneration. Hepatology 2021, 74, 973–986. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 367–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocca, C.; Protopapa, F.; Foglia, B.; Maggiora, M.; Cannito, S.; Parola, M.; Novo, E. Hepatic Myofibroblasts: A Heterogeneous and Redox-Modulated Cell Population in Liver Fibrogenesis. Antioxidants 2022, 11, 1278. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Bailey, S.M. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxid. Redox Signal. 2011, 15, 421–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- Loureiro, D.; Tout, I.; Narguet, S.; Bed, C.M.; Roinard, M.; Sleiman, A.; Boyer, N.; Pons-Kerjean, N.; Castelnau, C.; Giuly, N.; et al. Mitochondrial stress in advanced fibrosis and cirrhosis associated with chronic hepatitis B, chronic hepatitis C, or nonalcoholic steatohepatitis. Hepatology 2023, 77, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Geng, J.; Li, L.; Feng, X. An overview of ferroptosis in non-alcoholic fatty liver disease. Biomed. Pharmacother. 2022, 153, 113374. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, K.; Pinzani, M. Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents. Adv. Drug Deliv. Rev. 2017, 121, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; Ruart, M.; López-Sanjurjo, C.I.; Murphy, M.P.; Deulofeu, R.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; et al. Mitochondria-targeted antioxidant mitoquinone deactivates human and rat hepatic stellate cells and reduces portal hypertension in cirrhotic rats. Liver Int. 2017, 37, 1002–1012. [Google Scholar] [CrossRef]
- Sasaki, Y.; Dehnad, A.; Fish, S.; Sato, A.; Jiang, J.; Tian, J.; Schröder, K.; Brandes, R.; Török, N.J. NOX4 Regulates CCR2 and CCL2 mRNA Stability in Alcoholic Liver Disease. Sci. Rep. 2017, 7, 46144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andueza, A.; Garde, N.; García-Garzón, A.; Ansorena, E.; López-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martínez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease-novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Rantakari, P.; Patten, D.A.; Valtonen, J.; Karikoski, M.; Gerke, H.; Dawes, H.; Laurila, J.; Ohlmeier, S.; Elima, K.; Hübscher, S.G.; et al. Stabilin-1 expression defines a subset of macrophages that mediate tissue homeostasis and prevent fibrosis in chronic liver injury. Proc. Natl. Acad. Sci. USA 2016, 113, 9298–9303. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e6. [Google Scholar] [CrossRef] [Green Version]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Pinzani, M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 2022, 75, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Francque, S.; Sanyal, A. Breakthroughs in therapies for NASH and remaining challenges. J. Hepatol. 2022, 76, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Novo, E.; Parola, M. Therapeutic pro-fibrogenic signaling pathways in fibroblasts. Adv. Drug Deliv. Rev. 2017, 121, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Natarajan, S.K.; Chung, S. Gamma-Tocotrienol Attenuates the Hepatic Inflammation and Fibrosis by Suppressing Endoplasmic Reticulum Stress in Mice. Mol. Nutr. Food Res. 2018, 62, e1800519. [Google Scholar] [CrossRef]
- Turkseven, S.; Bolognesi, M.; Brocca, A.; Pesce, P.; Angeli, P.; Di Pascoli, M. Mitochondria-targeted antioxidant mitoquinone attenuates liver inflammation and fibrosis in cirrhotic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G298–G304. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Seen, S. Chronic liver disease and oxidative stress—A narrative review. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141, Erratum in Free Radic. Biol. Med. 2021, 162, 174. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeon, J.H. Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 2846. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Seedorf, K.; Weber, C.; Vinson, C.; Berger, S.; Vuillard, L.M.; Kiss, A.; Creusot, S.; Broux, O.; Geant, A.; Ilic, C.; et al. Selective disruption of NRF2-KEAP1 interaction leads to NASH resolution and reduction of liver fibrosis in mice. JHEP Rep. 2022, 5, 100651. [Google Scholar] [CrossRef] [PubMed]
- Shu, G.; Yusuf, A.; Dai, C.; Sun, H.; Deng, X. Piperine inhibits AML-12 hepatocyte EMT and LX-2 HSC activation and alleviates mouse liver fibrosis provoked by CCl4: Roles in the activation of the Nrf2 cascade and subsequent suppression of the TGF-β1/Smad axis. Food Funct. 2021, 12, 11686–11703. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. NOX as a Therapeutic Target in Liver Disease. Antioxidants 2022, 11, 2038. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Garzarella, J.; Charlton, A.; Urner, S.; Østergaard, J.A.; Okabe, J.; Holterman, C.E.; Skene, A.; Power, D.A.; et al. Independent of Renox, NOX5 Promotes Renal Inflammation and Fibrosis in Diabetes by Activating ROS-Sensitive Pathways. Diabetes 2022, 71, 1282–1298. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Cheng, J.; Thannickal, V.J. Targeting NOX enzymes in pulmonary fibrosis. Cell. Mol. Life Sci. 2012, 69, 2365–2371. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Jandeleit-Dahm, K.; Szyndralewiez, C.; Török, N.J. Pre-clinical evidence of a dual NADPH oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung fibrosis. J. Cell. Mol. Med. 2023, 27, 471–481. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schröder, K.; Brandes, R.P.; Devaraj, S.; Török, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Bai, Y.; He, S.; Sun, S.; Jiang, X.; Yang, Z.; Lu, D.; Wei, P.; Liang, Y.; Peng, C.; et al. Sirtuin 1 ameliorates defenestration in hepatic sinusoidal endothelial cells during liver fibrosis via inhibiting stress-induced premature senescence. Cell Prolif. 2021, 54, e12991. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.K.S.; Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 2018, 75, 2951–2961. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.C.; Bargut, T.C.; de Carvalho, S.N.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Peroxisome proliferator-activated receptors-alpha and gamma are targets to treat offspring from maternal diet-induced obesity in mice. PLoS ONE 2013, 8, e64258. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clarià, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Kong, D.; Lu, Y.; Zheng, S. Peroxisome proliferator-activated receptor-γ as a therapeutic target for hepatic fibrosis: From bench to bedside. Cell. Mol. Life Sci. 2013, 70, 259–276. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hong, B.; Bian, M.; Jin, H.; Chen, J.; Shao, J.; Zhang, F.; Zheng, S. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a PPARγ-dependent manner. Int. Immunopharmacol. 2019, 75, 105816. [Google Scholar] [CrossRef]
- Chen, H.; Tan, H.; Wan, J.; Zeng, Y.; Wang, J.; Wang, H.; Lu, X. PPAR-γ signaling in nonalcoholic fatty liver disease: Pathogenesis and therapeutic targets. Pharmacol. Ther. 2023, 245, 108391. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Nagata, K. Redox-Mediated Regulatory Mechanisms of Endoplasmic Reticulum Homeostasis. Cold Spring Harb. Perspect. Biol. 2019, 11, a033910. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, B.; Meng, X.; Yao, S.; Jin, L.; Yang, J.; Wang, J.; Zhang, H.; Zhang, Z.; Cai, D.; et al. Berberine prevents progression from hepatic steatosis to steatohepatitis and fibrosis by reducing endoplasmic reticulum stress. Sci. Rep. 2016, 6, 20848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Martin-Gronert, M.S.; McConnell, J.M.; Ozanne, S.E. Coenzyme Q10 prevents hepatic fibrosis, inflammation, and oxidative stress in a male rat model of poor maternal nutrition and accelerated postnatal growth. Am. J. Clin. Nutr. 2016, 103, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.K.; Pokharel, Y.R.; Lim, S.C.; Han, H.K.; Ryu, C.S.; Kim, S.K.; Kwak, M.K.; Kang, K.W. Inhibition of liver fibrosis by solubilized coenzyme Q10: Role of Nrf2 activation in inhibiting transforming growth factor-beta1 expression. Toxicol. Appl. Pharmacol. 2009, 240, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.; Liu, Q.; Krishnasamy, Y.; Shi, Z.; Ramshesh, V.K.; Haque, K.; Schnellmann, R.G.; Murphy, M.P.; Lemasters, J.J.; Rockey, D.C.; et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 14–27. [Google Scholar] [PubMed]
- Dou, S.D.; Zhang, J.N.; Xie, X.L.; Liu, T.; Hu, J.L.; Jiang, X.Y.; Wang, M.M.; Jiang, H.D. MitoQ inhibits hepatic stellate cell activation and liver fibrosis by enhancing PINK1/parkin-mediated mitophagy. Open Med. 2021, 16, 1718–1727. [Google Scholar] [CrossRef]
- Shan, S.; Liu, Z.; Liu, Z.; Zhang, C.; Song, F. MitoQ alleviates carbon tetrachloride-induced liver fibrosis in mice through regulating JNK/YAP pathway. Toxicol. Res. 2022, 11, 852–862. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Qiao, L.; Guo, Z.; Liu, H.; Liu, J.; Lin, X.; Deng, H.; Liu, X.; Zhao, Y.; Xiao, X.; Lei, J.; et al. Protective Effect of Mitophagy Regulated by mTOR Signaling Pathway in Liver Fibrosis Associated with Selenium. Nutrients 2022, 14, 2410. [Google Scholar] [CrossRef]
- Kang, J.W.; Hong, J.M.; Lee, S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016, 60, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, P.; Wang, J.; Toan, S.; Ren, J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct. Target. Ther. 2019, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, G.; Wang, J.; Deng, M.; Yuan, F.; Gong, J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020, 53, e12731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bu, Q.; Liu, M.; Zhang, R.; Gu, J.; Li, L.; Zhou, J.; Liang, Y.; Su, W.; Liu, Z.; et al. XBP1-mediated activation of the STING signalling pathway in macrophages contributes to liver fibrosis progression. JHEP Rep. 2022, 4, 100555. [Google Scholar] [CrossRef]
- Kim, K.M.; Cho, S.S.; Ki, S.H. Emerging roles of ferroptosis in liver pathophysiology. Arch. Pharmacal Res. 2020, 43, 985–996. [Google Scholar] [CrossRef]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The emerging role of ferroptosis in non-cancer liver diseases: Hype or increasing hope? Cell Death Dis. 2020, 11, 518. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, E.; Hu, H. Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies. Biomedicines 2021, 9, 1660. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81, Erratum in Am. J. Pathol. 2020, 190, 723. [Google Scholar] [CrossRef]
- Shi, J.F.; Liu, Y.; Wang, Y.; Gao, R.; Wang, Y.; Liu, J. Targeting ferroptosis, a novel programmed cell death, for the potential of alcohol-related liver disease therapy. Front. Pharmacol. 2023, 14, 1194343. [Google Scholar] [CrossRef]
- Pan, Q.; Luo, Y.; Xia, Q.; He, K. Ferroptosis and Liver Fibrosis. Int. J. Med. Sci. 2021, 18, 3361–3366. [Google Scholar] [CrossRef]
- Zhu, L.; Luo, S.; Zhu, Y.; Tang, S.; Li, C.; Jin, X.; Wu, F.; Jiang, H.; Wu, L.; Xu, Y. The Emerging Role of Ferroptosis in Various Chronic Liver Diseases: Opportunity or Challenge. J. Inflamm. Res. 2023, 16, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Wu, S.; Tan, S.; Qin, Y.; Wang, X.; Jiang, J.; Liu, H.; Wu, B. Berberine alleviates liver fibrosis through inducing ferrous redox to activate ROS-mediated hepatic stellate cells ferroptosis. Cell Death Discov. 2021, 7, 374. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Ishida, K.; Kaji, K.; Sato, S.; Ogawa, H.; Takagi, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane ameliorates ethanol plus carbon tetrachloride-induced liver fibrosis in mice through the Nrf2-mediated antioxidant response and acetaldehyde metabolization with inhibition of the LPS/TLR4 signaling pathway. J. Nutr. Biochem. 2021, 89, 108573. [Google Scholar] [CrossRef]
- Hurtado-Navarro, L.; Angosto-Bazarra, D.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. NLRP3 Inflammasome and Pyroptosis in Liver Pathophysiology: The Emerging Relevance of Nrf2 Inducers. Antioxidants 2022, 11, 870. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blas-García, A.; Apostolova, N. Novel Therapeutic Approaches to Liver Fibrosis Based on Targeting Oxidative Stress. Antioxidants 2023, 12, 1567. https://doi.org/10.3390/antiox12081567
Blas-García A, Apostolova N. Novel Therapeutic Approaches to Liver Fibrosis Based on Targeting Oxidative Stress. Antioxidants. 2023; 12(8):1567. https://doi.org/10.3390/antiox12081567
Chicago/Turabian StyleBlas-García, Ana, and Nadezda Apostolova. 2023. "Novel Therapeutic Approaches to Liver Fibrosis Based on Targeting Oxidative Stress" Antioxidants 12, no. 8: 1567. https://doi.org/10.3390/antiox12081567
APA StyleBlas-García, A., & Apostolova, N. (2023). Novel Therapeutic Approaches to Liver Fibrosis Based on Targeting Oxidative Stress. Antioxidants, 12(8), 1567. https://doi.org/10.3390/antiox12081567