
Desmin Reorganization by Stimuli Inducing Oxidative Stress and Electrophiles: Role of Its Single Cysteine Residue

 ,
,  and
and 
Abstract
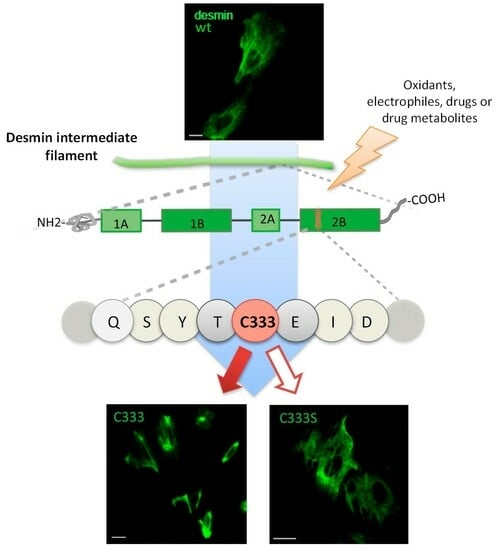
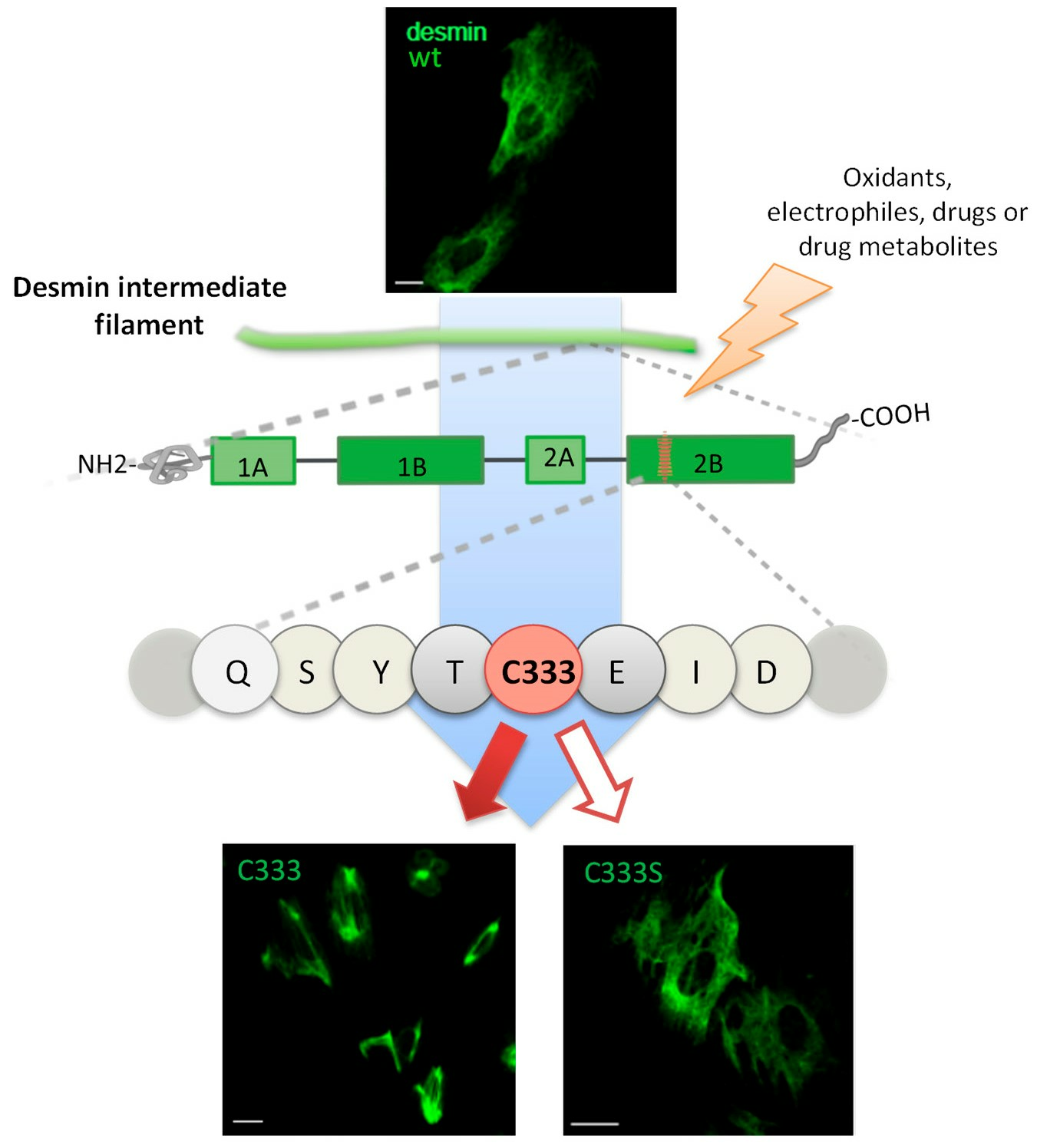
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
3.1. Cys333 of Desmin Is a Selective Target for Oxidants and Electrophiles In Vitro
3.2. H2O2 Disrupts Cellular Desmin Organization and This Is Attenuated in a C333S Desmin Mutant
3.3. 15d-PGJ2 Elicits Desmin Network Alterations, Which Are More Intense in the wt Protein
3.4. Chemical Hypoxia Elicits Desmin Rearrangement Dependent on C333
3.5. Importance of C333 for Desmin Organization in Cells under Non-Stress Conditions
4. Discussion
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.-M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Pallari, H.-M.; Eriksson, J.E. Intermediate filaments as signaling platforms. Sci. STKE 2006, 2006, pe53. [Google Scholar] [CrossRef]
- Toivola, D.M.; Strnad, P.; Habtezion, A.; Omary, M.B. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010, 20, 79–91. [Google Scholar] [CrossRef]
- Palmisano, M.G.; Bremner, S.N.; Hornberger, T.A.; Meyer, G.A.; Domenighetti, A.A.; Shah, S.B.; Kiss, B.; Kellermayer, M.; Ryan, A.F.; Lieber, R.L. Skeletal muscle intermediate filaments form a stress-transmitting and stress-signaling network. J. Cell Sci. 2015, 128, 219–224. [Google Scholar]
- Viedma-Poyatos, Á.; Pajares, M.A.; Pérez-Sala, D. Type III intermediate filaments as targets and effectors of electrophiles and oxidants. Redox Biol. 2020, 36, 101582. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of vimentin in health and disease. Genes Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- González-Jiménez, P.; Duarte, S.; Martínez-Fernández, A.; Navarro-Carrasco, E.; Lalioti, V.; Pajares, M.A.; Pérez-Sala, D. Vimentin single cysteine residue acts as a tunable sensor for network organization and as a key for actin remodeling in response to oxidants and electrophiles. Redox Biol. 2023, 64, 102756. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Dalakas, M.C. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Investig. 2009, 119, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Schaart, G.; Viebahn, C.; Langmann, W.; Ramaekers, F. Desmin and titin expression in early postimplantation mouse embryos. Development 1989, 107, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Milner, D.J.; Weitzer, G. Desmin in muscle formation and maintenance: Knockouts and consequences. Cell Struct. Funct. 1997, 22, 103–116. [Google Scholar] [CrossRef]
- Thornell, L.; Carlsson, L.; Li, Z.; Mericskay, M.; Paulin, D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J. Mol. Cell. Cardiol. 1997, 29, 2107–2124. [Google Scholar] [CrossRef]
- Li, Z.; Colucci-Guyon, E.; Pinçon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef]
- Mavroidis, M.; Capetanaki, Y. Extensive induction of important mediators of fibrosis and dystrophic calcification in desmin-deficient cardiomyopathy. Am. J. Pathol. 2002, 160, 943–952. [Google Scholar] [CrossRef]
- Singh, S.R.; Kadioglu, H.; Patel, K.; Carrier, L.; Agnetti, G. Is Desmin Propensity to Aggregate Part of its Protective Function? Cells 2020, 9, 491. [Google Scholar] [CrossRef]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef]
- Fountoulakis, M.; Soumaka, E.; Rapti, K.; Mavroidis, M.; Tsangaris, G.; Maris, A.; Weisleder, N.; Capetanaki, Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J. Mol. Cell. Cardiol. 2005, 38, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free. Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef]
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schröder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bryson, V.G.; Wang, C.; Li, T.; Kerr, J.P.; Wilson, R.; Muoio, D.M.; Bloch, R.J.; Ward, C.; Rosenberg, P.B. Desmin interacts with STIM1 and coordinates Ca2+ signaling in skeletal muscle. J. Clin. Investig. 2021, 6, e143472. [Google Scholar] [CrossRef]
- Dayal, A.A.; Medvedeva, N.V.; Nekrasova, T.M.; Duhalin, S.D.; Surin, A.K.; Minin, A.A. Desmin Interacts Directly with Mitochondria. Int. J. Mol. Sci. 2020, 21, 8122. [Google Scholar] [CrossRef]
- Agnetti, G.; Herrmann, H.; Cohen, S. New roles for desmin in the maintenance of muscle homeostasis. FEBS J. 2022, 289, 2755–2770. [Google Scholar] [CrossRef]
- Delort, F.; Segard, B.-D.; Hakibilen, C.; Bourgois-Rocha, F.; Cabet, E.; Vicart, P.; Huang, M.-E.; Clary, G.; Lilienbaum, A.; Agbulut, O.; et al. Alterations of redox dynamics and desmin post-translational modifications in skeletal muscle models of desminopathies. Exp. Cell Res. 2019, 383, 111539. [Google Scholar] [CrossRef]
- Janué, A.; Odena, M.A.; Oliveira, E.; Olivé, M.; Ferrer, I. Desmin is oxidized and nitrated in affected muscles in myotilinopathies and desminopathies. J. Neuropathol. Exp. Neurol. 2007, 66, 711–723. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, J.; Huang, F.; Huang, M.; Zhou, G. Influence of oxidation on the susceptibility of purified desmin to degradation by mu-calpain, caspase-3 and -6. Food Chem. 2014, 150, 220–226. [Google Scholar] [CrossRef]
- Segard, B.-D.; Delort, F.; Bailleux, V.; Simon, S.; Leccia, E.; Gausseres, B.; Briki, F.; Vicart, P.; Batonnet-Pichon, S. N-acetyl-L-cysteine prevents stress-induced desmin aggregation in cellular models of desminopathy. PLoS ONE 2013, 8, e76361. [Google Scholar] [CrossRef]
- Cabet, E.; Batonnet-Pichon, S.; Delort, F.; Gausserès, B.; Vicart, P.; Lilienbaum, A. Antioxidant Treatment and Induction of Autophagy Cooperate to Reduce Desmin Aggregation in a Cellular Model of Desminopathy. PLoS ONE 2015, 10, e0137009. [Google Scholar] [CrossRef]
- Guichard, J.L.; Rogowski, M.; Agnetti, G.; Fu, L.; Powell, P.; Wei, C.-C.; Collawn, J.; Dell’italia, L.J. Desmin loss and mitochondrial damage precede left ventricular systolic failure in volume overload heart failure. Am. J. Physiol. Circ. Physiol. 2017, 313, H32–H45. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Mechref, Y.; Marini, M.; Tonin, P.; Minuz, P.; Grigoli, L.; Guglielmi, V.; Klouckova, I.; Chiamulera, C.; Meneguzzi, A.; et al. Increased protein nitration in mitochondrial diseases: Evidence for vessel wall involvement. Mol. Cell. Proteom. 2011, 10, M110.002964. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Poyatos, Á.; González-Jiménez, P.; Langlois, O.; Company-Marín, I.; Spickett, C.M.; Pérez-Sala, D. Protein Lipoxidation: Basic Concepts and Emerging Roles. Antioxidants 2021, 10, 295. [Google Scholar] [CrossRef]
- Luo, J.; Hill, B.G.; Gu, Y.; Cai, J.; Srivastava, S.; Bhatnagar, A.; Prabhu, S.D. Mechanisms of acrolein-induced myocardial dysfunction: Implications for environmental and endogenous aldehyde exposure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3673–H3684. [Google Scholar] [CrossRef]
- Caruso, G.; Privitera, A.; Antunes, B.M.; Lazzarino, G.; Lunte, S.M.; Aldini, G.; Caraci, F. The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway. Molecules 2022, 27, 4452. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Oeste, C.L.; Martínez, A.E.; Garzón, B.; Carrasco, M.J.; Cañada, F.J. Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat. Commun. 2015, 6, 7287. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Poyatos, Á.; de Pablo, Y.; Pekny, M.; Pérez-Sala, D. The cysteine residue of glial fibrillary acidic protein is a critical target for lipoxidation and required for efficient network organization. Free. Radic. Biol. Med. 2018, 120, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Mónico, A.; Duarte, S.; Pajares, M.A.; Pérez-Sala, D. Vimentin disruption by lipoxidation and electrophiles: Role of the cysteine residue and filament dynamics. Redox Biol. 2019, 23, 101098. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.A.; Franke, W.W. Heteropolymer filaments of vimentin and desmin in vascular smooth muscle tissue and cultured baby hamster kidney cells demonstrated by chemical crosslinking. Proc. Natl. Acad. Sci. USA 1982, 79, 3452–3456. [Google Scholar] [CrossRef]
- Canton, M.; Neverova, I.; Menabò, R.; Van Eyk, J.; Di Lisa, F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am. J. Physiol. Circ. Physiol. 2004, 286, H870–H877. [Google Scholar] [CrossRef]
- Li, Z.; Jun, S.; Singh, K.K.; Calhoun, P.J.; Keceli, G.; Patel, K.; Kadioglu, H.; Paolocci, N.; Agnetti, G. Ischemia/Reperfusion Injury and Oxidative Stress Impair Cardiac Desmin Proteostasis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Brennan, J.P.; Wait, R.; Begum, S.; Bell, J.R.; Dunn, M.J.; Eaton, P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J. Biol. Chem. 2004, 279, 41352–41360. [Google Scholar] [CrossRef]
- Charles, R.L.; Schroder, E.; May, G.; Free, P.; Gaffney, P.R.; Wait, R.; Begum, S.; Heads, R.J.; Eaton, P. Protein sulfenation as a redox sensor: Proteomics studies using a novel biotinylated dimedone analogue. Mol. Cell. Proteom. 2007, 6, 1473–1484. [Google Scholar] [CrossRef]
- VanHecke, G.C.; Abeywardana, M.Y.; Huang, B.; Ahn, Y. Isotopically Labeled Clickable Glutathione to Quantify Protein S-Glutathionylation. ChemBioChem 2020, 21, 853–859. [Google Scholar] [CrossRef]
- Abeywardana, M.Y.; Samarasinghe, K.T.G.; Godage, D.M.; Ahn, Y.-H. Identification and Quantification of Glutathionylated Cysteines under Ischemic Stress. J. Proteome Res. 2021, 20, 4529–4542. [Google Scholar] [CrossRef]
- Mónico, A.; Rodríguez-Senra, E.; Cañada, F.J.; Zorrilla, S.; Pérez-Sala, D. Drawbacks of dialysis procedures for removal of EDTA. PLoS ONE 2017, 12, e0169843. [Google Scholar] [CrossRef] [PubMed]
- Mónico, A.; Zorrilla, S.; Rivas, G.; Pérez-Sala, D. Zinc Differentially Modulates the Assembly of Soluble and Polymerized Vimentin. Int. J. Mol. Sci. 2020, 21, 2426. [Google Scholar] [CrossRef]
- Mónico, A.; Guzmán-Caldentey, J.; Pajares, M.A.; Martín-Santamaría, S.; Pérez-Sala, D. Molecular Insight into the Regulation of Vimentin by Cysteine Modifications and Zinc Binding. Antioxidants 2021, 10, 1039. [Google Scholar] [CrossRef]
- Sarria, A.; Lieber, J.; Nordeen, S.; Evans, R. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J. Cell Sci. 1994, 107 Pt 6, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V.; González-Sanz, S.; Lois-Bermejo, I.; González-Jiménez, P.; Viedma-Poyatos, Á.; Merino, A.; Pajares, M.A.; Pérez-Sala, D. Cell surface detection of vimentin, ACE2 and SARS-CoV-2 Spike proteins reveals selective colocalization at primary cilia. Sci. Rep. 2022, 12, 7063. [Google Scholar] [CrossRef] [PubMed]
- Lois-Bermejo, I.; González-Jiménez, P.; Duarte, S.; Pajares, M.A.; Pérez-Sala, D. Vimentin tail segments are differentially exposed at distinct cellular locations and in response to stress. Front. Cell Dev. Biol. 2022, 10, 908263. [Google Scholar] [CrossRef]
- Viedma-Poyatos, Á.; González-Jiménez, P.; Pajares, M.A.; Pérez-Sala, D. Alexander disease GFAP R239C mutant shows increased susceptibility to lipoxidation and elicits mitochondrial dysfunction and oxidative stress. Redox Biol. 2022, 55, 102415. [Google Scholar] [CrossRef]
- Rogers, K.R.; Herrmann, H.; Franke, W.W. Characterization of disulfide crosslink formation of human vimentin at the dimer, tetramer, and intermediate filament levels. J. Struct. Biol. 1996, 117, 55–69. [Google Scholar] [CrossRef]
- Oeste, C.L.; Pérez-Sala, D. Modification of cysteine residues by cyclopentenone prostaglandins: Interplay with redox regulation of protein function. Mass Spectrom. Rev. 2014, 33, 110–125. [Google Scholar] [CrossRef]
- Sanchez-Gomez, F.J.; Dorado, C.G.; Ayuso, P.; Agundez, J.A.; Pajares, M.A.; Perez-Sala, D. Modulation of GSTP1-1 oligomerization by inflammatory mediators and reactive drugs. Inflamm. Allergy-Drug Targets 2013, 12, 162–171. [Google Scholar] [CrossRef]
- Gonzalez-Morena, J.; Montanez, M.; Aldini, G.; Sanchez-Gomez, F.; Perez-Sala, D. Adduct formation and context factors in drug hypersensitivity: Insight from proteomic studies. Curr. Pharm. Des. 2016, 22, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.A.; Isin, E.M.; Ogese, M.O.; Mettetal, J.T.; Williams, D.P. Reactive Metabolites: Current and Emerging Risk and Hazard Assessments. Chem. Res. Toxicol. 2016, 29, 505–533. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustín, P.; Izquierdo-Álvarez, A.; Sánchez-Gómez, F.J.; Ramos, E.; Villa-Piña, T.; Lamas, S.; Bogdanova, A.; Martínez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free. Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, W. Regulation of Endothelial Progenitor Cell Functions in Ischemic Heart Disease: New Therapeutic Targets for Cardiac Remodeling and Repair. Front. Cardiovasc. Med. 2022, 9, 896782. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- Cervellati, F.; Cervellati, C.; Romani, A.; Cremonini, E.; Sticozzi, C.; Belmonte, G.; Pessina, F.; Valacchi, G. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free. Radic. Res. 2014, 48, 303–312. [Google Scholar] [CrossRef]
- Semenza, G.; Roth, P.; Fang, H.; Wang, G. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Beru, N.; McDonald, J.; Lacombe, C.; Goldwasser, E. Expression of the erythropoietin gene. Mol. Cell. Biol. 1986, 6, 2571–2575. [Google Scholar]
- Lendahl, U.; Lee, K.L.; Yang, H.; Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 2009, 10, 821–832. [Google Scholar] [CrossRef]
- Minchenko, A.; Caro, J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: Role of hypoxia responsive element. Mol. Cell. Biochem. 2000, 208, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.L.; Paulin, D.; Mericskay, M.; Li, Z. Posttranslational modifications of desmin and their implication in biological processes and pathologies. Histochem. Cell Biol. 2014, 141, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Aldini, G.; Ito, S.; Morishita, N.; Shibata, T.; Vistoli, G.; Carini, M.; Uchida, K. Δ12-Prostaglandin J2 as a Product and Ligand of Human Serum Albumin: Formation of an Unusual Covalent Adduct at His146. J. Am. Chem. Soc. 2010, 132, 824–832. [Google Scholar] [CrossRef]
- Oeste, C.L.; Díez-Dacal, B.; Bray, F.; de Lacoba, M.G.; de la Torre, B.G.; Andreu, D.; Ruiz-Sánchez, A.J.; Pérez-Inestrosa, E.; García-Domínguez, C.A.; Rojas, J.M.; et al. The C-terminus of H-Ras as a target for the covalent binding of reactive compounds modulating Ras-dependent pathways. PLoS ONE 2011, 6, e15866. [Google Scholar] [CrossRef] [PubMed]
- Ploemen, J.H.; Van Schanke, A.; Van Ommen, B.; Van Bladeren, P.J. Reversible conjugation of ethacrynic acid with glutathione and human glutathione S-transferase P1-1. Cancer Res. 1994, 54, 915–919. [Google Scholar] [PubMed]
- Leeming, M.G.; Gamon, L.F.; Wille, U.; Donald, W.A.; O’Hair, R.A. What Are the Potential Sites of Protein Arylation by N-Acetyl-p-benzoquinone Imine (NAPQI)? Chem. Res. Toxicol. 2015, 28, 2224–2233. [Google Scholar] [CrossRef]
- Bessho, M.; Aki, T.; Funakoshi, T.; Unuma, K.; Noritake, K.; Kato, C.; Uemura, K. Rho-kinase inhibitor Y-27632 attenuates arsenic trioxide toxicity in H9c2 cardiomyoblastoma cells. Cardiovasc. Toxicol. 2013, 13, 267–277. [Google Scholar] [CrossRef]
- Boccellino, M.; Galasso, G.; Ambrosio, P.; Stiuso, P.; Lama, S.; Di Zazzo, E.; Schiavon, S.; Vecchio, D.; D’ambrosio, L.; Quagliuolo, L.; et al. H9c2 Cardiomyocytes under Hypoxic Stress: Biological Effects Mediated by Sentinel Downstream Targets. Oxidative Med. Cell. Longev. 2021, 2021, 6874146. [Google Scholar] [CrossRef]
- Djabali, K.; Piron, G.; de Néchaud, B.; Portier, M.-M. αB-crystallin interacts with cytoplasmic intermediate filament bundles during mitosis. Exp. Cell Res. 1999, 253, 649–662. [Google Scholar] [CrossRef]
- Der Perng, M.; Cairns, L.; Ijssel, P.v.D.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate filament interactions can be altered by HSP27 and αB-crystallin. J. Cell Sci. 1999, 112 Pt 13, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Giles, G.; Jacob, C. Reactive sulfur species: An emerging concept in oxidative stress. Biol. Chem. 2002, 383, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Iciek, M.; Kowalczyk-Pachel, D.; Bilska-Wilkosz, A.; Kwiecień, I.; Górny, M.; Włodek, L. S-sulfhydration as a cellular redox regulation. Biosci. Rep. 2016, 36, e00304. [Google Scholar] [CrossRef] [PubMed]
- Rainer, P.P.; Dong, P.; Sorge, M.; Fert-Bober, J.; Holewinski, R.J.; Wang, Y.; Foss, C.A.; An, S.S.; Baracca, A.; Solaini, G.; et al. Desmin Phosphorylation Triggers Preamyloid Oligomers Formation and Myocyte Dysfunction in Acquired Heart Failure. Circ. Res. 2018, 122, e75–e83. [Google Scholar] [CrossRef]
- Kedia, N.; Arhzaouy, K.; Pittman, S.K.; Sun, Y.; Batchelor, M.; Weihl, C.C.; Bieschke, J. Desmin forms toxic, seeding-competent amyloid aggregates that persist in muscle fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 16835–16840. [Google Scholar] [CrossRef]
- Agnetti, G.; Halperin, V.L.; Kirk, J.A.; Chakir, K.; Guo, Y.; Lund, L.; Nicolini, F.; Gherli, T.; Guarnieri, C.; Caldarera, C.M.; et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc. Res. 2014, 102, 24–34. [Google Scholar] [CrossRef]
- Pekny, M.; Lane, E.B. Intermediate filaments and stress. Exp. Cell Res. 2007, 313, 2244–2254. [Google Scholar] [CrossRef]
- Parvanian, S.; Zha, H.; Su, D.; Xi, L.; Jiu, Y.; Chen, H.; Eriksson, J.E.; Cheng, F. Exosomal Vimentin from Adipocyte Progenitors Protects Fibroblasts against Osmotic Stress and Inhibits Apoptosis to Enhance Wound Healing. Int. J. Mol. Sci. 2021, 22, 4678. [Google Scholar] [CrossRef]
- Pogoda, K.; Byfield, F.; Deptuła, P.; Cieśluk, M.; Suprewicz, Ł.; Skłodowski, K.; Shivers, J.L.; van Oosten, A.; Cruz, K.; Tarasovetc, E.; et al. Unique Role of Vimentin Networks in Compression Stiffening of Cells and Protection of Nuclei from Compressive Stress. Nano Lett. 2022, 22, 4725–4732. [Google Scholar] [CrossRef]
- Quinlan, R. Cytoskeletal competence requires protein chaperones. Prog. Mol. Subcell. Biol. 2002, 28, 219–233. [Google Scholar]
- Pekovic, V.; Gibbs-Seymour, I.; Markiewicz, E.; Alzoghaibi, F.; Benham, A.M.; Edwards, R.; Wenhert, M.; von Zglinicki, T.; Hutchison, C.J. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011, 10, 1067–1079. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moneo-Corcuera, D.; Viedma-Poyatos, Á.; Stamatakis, K.; Pérez-Sala, D. Desmin Reorganization by Stimuli Inducing Oxidative Stress and Electrophiles: Role of Its Single Cysteine Residue. Antioxidants 2023, 12, 1703. https://doi.org/10.3390/antiox12091703
Moneo-Corcuera D, Viedma-Poyatos Á, Stamatakis K, Pérez-Sala D. Desmin Reorganization by Stimuli Inducing Oxidative Stress and Electrophiles: Role of Its Single Cysteine Residue. Antioxidants. 2023; 12(9):1703. https://doi.org/10.3390/antiox12091703
Chicago/Turabian StyleMoneo-Corcuera, Diego, Álvaro Viedma-Poyatos, Konstantinos Stamatakis, and Dolores Pérez-Sala. 2023. "Desmin Reorganization by Stimuli Inducing Oxidative Stress and Electrophiles: Role of Its Single Cysteine Residue" Antioxidants 12, no. 9: 1703. https://doi.org/10.3390/antiox12091703
APA StyleMoneo-Corcuera, D., Viedma-Poyatos, Á., Stamatakis, K., & Pérez-Sala, D. (2023). Desmin Reorganization by Stimuli Inducing Oxidative Stress and Electrophiles: Role of Its Single Cysteine Residue. Antioxidants, 12(9), 1703. https://doi.org/10.3390/antiox12091703