



High Sucrose Diet-Induced Subunit I Tyrosine 304 Phosphorylation of Cytochrome c Oxidase Leads to Liver Mitochondrial Respiratory Dysfunction in the Cohen Diabetic Rat Model

 ,
,  , , ,
, , ,  and
and
Abstract
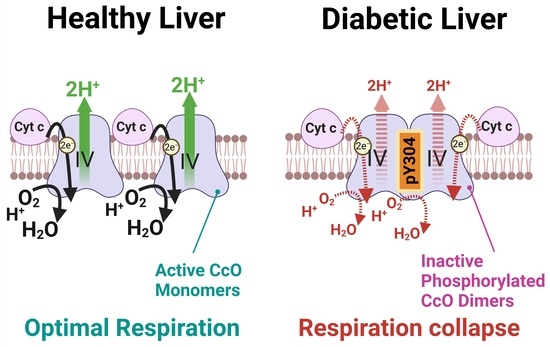
:
1. Introduction
2. Materials and Methods
2.1. Cohen Diabetes-Sensitive and Cohen Diabetes-Resistant Rats Tissue Harvesting
2.2. Liver Tissue Homogenization and Mitochondrial Enrichment for Blue Native PAGE (BN-PAGE)
2.3. Mitochondrial Membrane Solubilization for BN-PAGE
2.4. Mitochondrial Native Protein In-Gel Activity Assay (IGA)
2.5. Cytochrome c Oxidase Activity Measurements Using the Polarographic Method
2.6. Western Blot Analysis
2.7. ATP Measurements
2.8. Statistical Analyses
3. Results
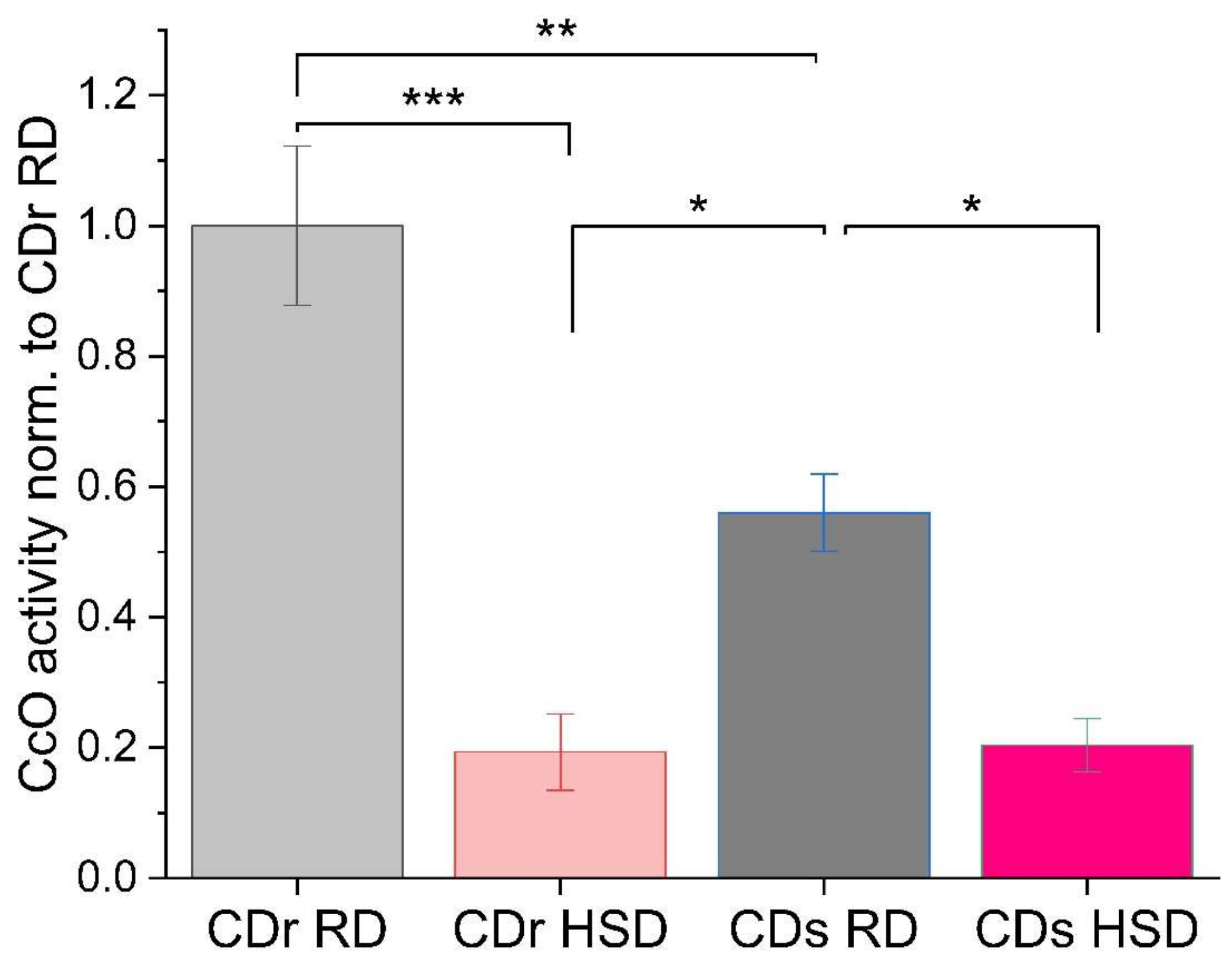
3.1. CcO Activity Is Decreased in CDr and CDs Rat Liver Mitochondria following HSD
3.2. Analysis of Electron Transport Chain Supercomplex Composition and Activity Reveal Pronounced Reduction of CcO Activity in the Dimeric Enzyme
3.3. High-Sucrose Diet Leads to Phosphorylation of CcO Subunit I Tyrosine 304 in the CcO Dimer
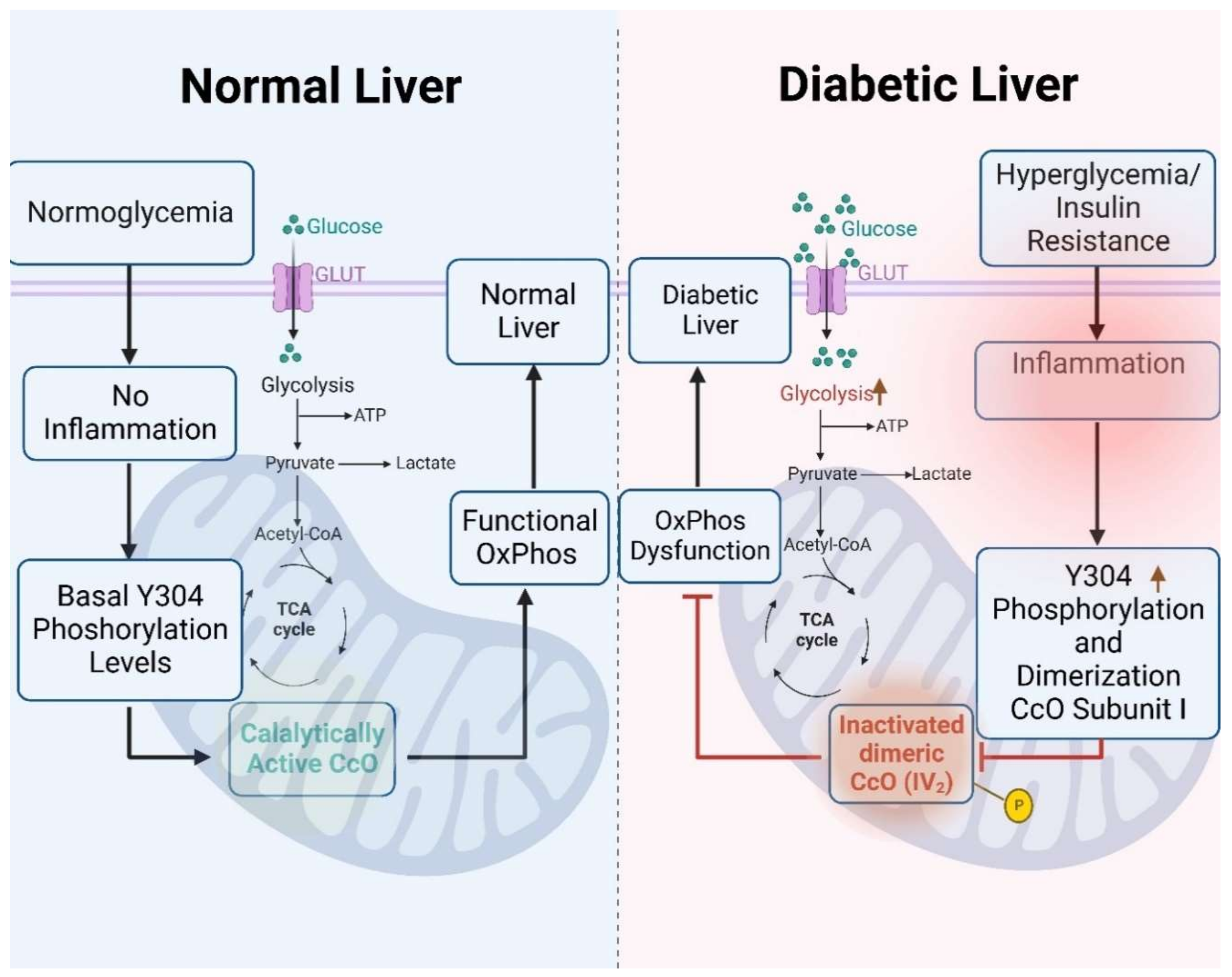
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications. Endocrinology 2020, 161, bqaa017. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spegel, P.; Mulder, H. The pathogenetic role of beta-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.M.; Teitelbaum, A.; Saliternik, R. Genetics and diet as factors in development of diabetes mellitus. Metabolism 1972, 21, 235–240. [Google Scholar] [CrossRef]
- Weksler-Zangen, S.; Yagil, C.; Zangen, D.H.; Ornoy, A.; Jacob, H.J.; Yagil, Y. The newly inbred cohen diabetic rat: A nonobese normolipidemic genetic model of diet-induced type 2 diabetes expressing sex differences. Diabetes 2001, 50, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Weksler-Zangen, S.; Raz, I.; Lenzen, S.; Jorns, A.; Ehrenfeld, S.; Amir, G.; Oprescu, A.; Yagil, Y.; Yagil, C.; Zangen, D.H.; et al. Impaired glucose-stimulated insulin secretion is coupled with exocrine pancreatic lesions in the Cohen diabetic rat. Diabetes 2008, 57, 279–287. [Google Scholar] [CrossRef]
- Aharon-Hananel, G.; Romero-Afrima, L.; Saada, A.; Mantzur, C.; Raz, I.; Weksler-Zangen, S. Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells. Cells 2022, 11, 929. [Google Scholar] [CrossRef]
- Zigmond, E.; Zangen, S.W.; Pappo, O.; Sklair-Levy, M.; Lalazar, G.; Zolotaryova, L.; Raz, I.; Ilan, Y. Beta-glycosphingolipids improve glucose intolerance and hepatic steatosis of the Cohen diabetic rat. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E72–E78. [Google Scholar] [CrossRef]
- Weksler-Zangen, S.; Aharon-Hananel, G.; Mantzur, C.; Aouizerat, T.; Gurgul-Convey, E.; Raz, I.; Saada, A. IL-1beta hampers glucose-stimulated insulin secretion in Cohen diabetic rat islets through mitochondrial cytochrome c oxidase inhibition by nitric oxide. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E648–E657. [Google Scholar] [CrossRef]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1166–1171. [Google Scholar] [CrossRef]
- Villani, G.; Greco, M.; Papa, S.; Attardi, G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998, 273, 31829–31836. [Google Scholar] [CrossRef]
- Kunz, W.S.; Kudin, A.; Vielhaber, S.; Elger, C.E.; Attardi, G.; Villani, G. Flux control of cytochrome c oxidase in human skeletal muscle. J. Biol. Chem. 2000, 275, 27741–27745. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic. Biol. Med. 2000, 29, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Kadenbach, B. The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosterically. FEBS Lett. 1999, 443, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxid. Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Lee, I.; Grossman, L.I.; Doan, J.W.; Sanderson, T.H. Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: Respiration, apoptosis, and human disease. Adv. Exp. Med. Biol. 2012, 748, 237–264. [Google Scholar] [CrossRef]
- Lee, I.; Hüttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef]
- Lee, I.; Salomon, A.R.; Ficarro, S.; Mathes, I.; Lottspeich, F.; Grossman, L.I.; Hüttemann, M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J. Biol. Chem. 2005, 280, 6094–6100. [Google Scholar] [CrossRef]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Hüttemann, M. Tumor necrosis factor α inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef]
- Schagger, H.; von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Jha, P.; Wang, X.; Auwerx, J. Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schagger, H. Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2006, 1757, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Boekema, E.J.; Braun, H.P. Supramolecular structure of the mitochondrial oxidative phosphorylation system. J. Biol. Chem. 2007, 282, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schagger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef]
- Tomah, S.; Alkhouri, N.; Hamdy, O. Nonalcoholic fatty liver disease and type 2 diabetes: Where do Diabetologists stand? Clin. Diabetes Endocrinol. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Alimujiang, M.; Yu, X.Y.; Yu, M.Y.; Hou, W.L.; Yan, Z.H.; Yang, Y.; Bao, Y.Q.; Yin, J. Enhanced liver but not muscle OXPHOS in diabetes and reduced glucose output by complex I inhibition. J. Cell Mol. Med. 2020, 24, 5758–5771. [Google Scholar] [CrossRef]
- Ferguson-Miller, S.; Brautigan, D.L.; Margoliash, E. Correlation of the kinetics of electron transfer activity of various eukaryotic cytochromes c with binding to mitochondrial cytochrome c oxidase. J. Biol. Chem. 1976, 251, 1104–1115. [Google Scholar] [CrossRef]
- Chiang Morales, M.D.; Chang, C.Y.; Le, V.L.; Huang, I.T.; Tsai, I.L.; Shih, H.J.; Huang, C.J. High-Fructose/High-Fat Diet Downregulates the Hepatic Mitochondrial Oxidative Phosphorylation Pathway in Mice Compared with High-Fat Diet Alone. Cells 2022, 11, 3425. [Google Scholar] [CrossRef]
- Szendroedi, J.; Chmelik, M.; Schmid, A.I.; Nowotny, P.; Brehm, A.; Krssak, M.; Moser, E.; Roden, M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009, 50, 1079–1086. [Google Scholar] [CrossRef]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssak, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Jha, D.; Mitra Mazumder, P. High fat diet administration leads to the mitochondrial dysfunction and selectively alters the expression of class 1 GLUT protein in mice. Mol. Biol. Rep. 2019, 46, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, H.; Xiang, R.; Yang, W.; Ye, J.; Yin, R.; Yang, J.; Chi, Y. ATP Secretion and Metabolism in Regulating Pancreatic Beta Cell Functions and Hepatic Glycolipid Metabolism. Front. Physiol. 2022, 13, 918042. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Weksler-Zangen, S. Is Type 2 Diabetes a Primary Mitochondrial Disorder? Cells 2022, 11, 1617. [Google Scholar] [CrossRef]
- Araujo, M.C.; Soczek, S.H.S.; Pontes, J.P.; Pinto, B.A.S.; Franca, L.M.; Soley, B.D.S.; Santos, G.S.; Saminez, W.F.S.; Fernandes, F.K.M.; Lima, J.; et al. Analysis of the Effect of the TRPC4/TRPC5 Blocker, ML204, in Sucrose-Induced Metabolic Imbalance. Pharmaceuticals 2023, 16, 1100. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | 2 h Post-OGTT (mmol/L) | Body Weight (g) |
|---|---|---|---|
| CDr RD | 5 | 7.13 ± 1.02 | 328.3 ± 7.8 |
| CDr HSD | 5 | 7.44 ± 0.18 | 292.3 ± 5.0 |
| CDs RD | 5 | 7.69 ± 0.1 | 306.5 ± 5.4 |
| CDs HSD | 5 | 16.3 ± 1.6 * | 301.0 ± 12.9 |
| Antibody | Dilution | Company | Product ID |
|---|---|---|---|
| Beta-actin | 1:2000 | Proteintech | 60008-1-Ig |
| VDAC1/2 | 1:5000 | Proteintech | 10866-1-AP |
| NDUFB6 | 1:5000 | Abcam | ab110244 |
| Core I | 1:3000 | Abcam | ab110252 |
| MT-CO1 (CcO I) | 1:5000 | Invitrogen | PA5-68016 |
| Custom-made pY304 CcO I | 1:3000 | Abgent | RB 96601 |
| Anti-rabbit IgG HRP-linked | 1:5000 | Cell Signaling | 7074S |
| Anti-mouse IgG HRP-linked | 1:5000 | Cell Signaling | 7076S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroum, T.; Pham, L.; Raisanen, T.E.; Morse, P.T.; Wan, J.; Bell, J.; Lax, R.; Saada, A.; Hüttemann, M.; Weksler-Zangen, S. High Sucrose Diet-Induced Subunit I Tyrosine 304 Phosphorylation of Cytochrome c Oxidase Leads to Liver Mitochondrial Respiratory Dysfunction in the Cohen Diabetic Rat Model. Antioxidants 2024, 13, 19. https://doi.org/10.3390/antiox13010019
Arroum T, Pham L, Raisanen TE, Morse PT, Wan J, Bell J, Lax R, Saada A, Hüttemann M, Weksler-Zangen S. High Sucrose Diet-Induced Subunit I Tyrosine 304 Phosphorylation of Cytochrome c Oxidase Leads to Liver Mitochondrial Respiratory Dysfunction in the Cohen Diabetic Rat Model. Antioxidants. 2024; 13(1):19. https://doi.org/10.3390/antiox13010019
Chicago/Turabian StyleArroum, Tasnim, Lucynda Pham, Taryn E. Raisanen, Paul T. Morse, Junmei Wan, Jamie Bell, Rachel Lax, Ann Saada, Maik Hüttemann, and Sarah Weksler-Zangen. 2024. "High Sucrose Diet-Induced Subunit I Tyrosine 304 Phosphorylation of Cytochrome c Oxidase Leads to Liver Mitochondrial Respiratory Dysfunction in the Cohen Diabetic Rat Model" Antioxidants 13, no. 1: 19. https://doi.org/10.3390/antiox13010019
APA StyleArroum, T., Pham, L., Raisanen, T. E., Morse, P. T., Wan, J., Bell, J., Lax, R., Saada, A., Hüttemann, M., & Weksler-Zangen, S. (2024). High Sucrose Diet-Induced Subunit I Tyrosine 304 Phosphorylation of Cytochrome c Oxidase Leads to Liver Mitochondrial Respiratory Dysfunction in the Cohen Diabetic Rat Model. Antioxidants, 13(1), 19. https://doi.org/10.3390/antiox13010019